A GeoTES experiment involving the Weber-Tensleep formation
This page describes a reactive-transport simulation of a GeoTES system in the Weber-Tensleep formation. The transport is handled using the PorousFlow module, while the Geochemistry module is used to simulate a heat exchanger and the geochemistry. The simulations are coupled together in an operator-splitting approach using MultiApps.
In Geological Thermal Energy Storage (GeoTES), hot fluid is pumped into an subsurface confined aquifer, stored there for some time, then pumped back again to produce electricity. Many different scenarios have been proposed, including different borehole geometries (used to pump the fluid), different pumping strategies and cyclicities (pumping down certain boreholes, up other boreholes, etc), different temperatures, different aquifer properties, etc. This page only describes the heating phase of a particularly simple set up.
Two wells (boreholes) are used.
The cold (production) well, which pumps aquifer water from the aquifer.
The hot (injection) well, which pumps hot fluid into the aquifer.
These are connected by a heat-exchanger, which heats the aquifer water produced from (1) and passes it to (2). This means the fluid pumped through the injection well is, in fact, heated aquifer water, which is important practically from a water-conservation standpoint, and to assess the geochemical aspects of circulating aquifer water through the GeoTES system.
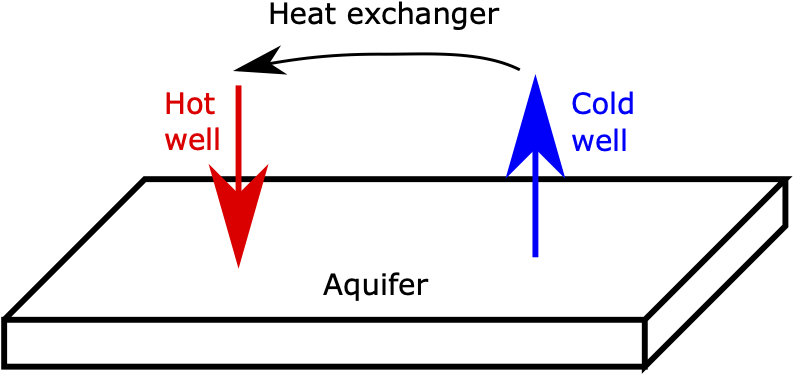
Figure 1: Schematic of the simple GeoTES simulation.
Observed geochemical composition and aquifer mineralogy
The composition of the Weber formation water has been measured at 25C and a typical analysis is shown in Table 1. Some minor points are:
Since HS is a redox species, its concentration fixes the oxidation state of the water (it is swapped with the basis species O(aq) in the simulations).
The species NH is not a basis species, but may be swapped into the basis to replace NO in the simulations.
The species B is not a basis species: B(OH) is used instead (with concentration 412mg.L).
The species Si is not a basis species: SiO(aq) is used instead (with concentration 96mg.L).
Table 1: Typical composition of the Weber formation water measured at 25C
| Species | Concentration (mg.L) |
|---|---|
| Cl- | 57400 |
| SO4– | 6030 |
| HCO3- | 3996 |
| HS- | 127 |
| Si | 45 |
| Al+++ | 3.5 |
| Ca++ | 539 |
| Mg++ | 45 |
| Fe++ | 44 |
| K+ | 1910 |
| Na+ | 36500 |
| Sr++ | 14 |
| F- | 6.1 |
| B | 72 |
| Br- | 99 |
| Ba++ | 14 |
| Li+ | 91 |
| NH3 (as N) | 33 |
| pH | 6.46 |
The mineralogy of the Weber-Tensleep aquifer has also been measured and a typical composition is shown in Table 2. The volume fractions do not sum to 100% because of small measurement errors.
Table 2: Typical mineralogy of the Weber-Tensleep aquifer
| Mineral | Volume fraction (%) |
|---|---|
| Quartz | 80.7 |
| K-feldspar | 8.0 |
| Kaolinite | 6.6E-05 |
| Siderite | 2.0 |
| Goethite | 0 |
| Pyrrhotite | 0.10 |
| Dolomite | 2.0 |
| Calcite | 5.0 |
| Fe-chlorite | 1.0 |
| Illite | 1.0 |
| Chalcedony | 0.11 |
| Anhydrite | 0.60 |
| Barite | 4.9E-05 |
| Celestite | 0 |
| Fluorite | 0 |
| Albite | 0 |
Equilibrium model at 25C
The simplest simulation possible is one that finds the molality of each primary and secondary species, given the measured concentrations of Table 1 while preventing any mineral precipitation. To perform this analysis, the concentrations shown in Table 1 must be converted to mole numbers, as shown in Table 3.
In Table 3 it is assumed that 1L of aqueous solution contains exactly 1kg of solvent water.
Table 3: Composition of the model at 25C. The pH is 6.46.
| Species | Measured conc (mg.L) | Mol weight (g/mol) | Molal (mol/kg(solvent water)) |
|---|---|---|---|
| Cl- | 57400 | 35.453 | 1.619044933 |
| SO4– | 6030 | 96.0576 | 0.062774835 |
| HCO3- | 3996 | 61.0171 | 0.065489838 |
| HS- | 127 | 33.0679 | 0.003840583 |
| SiO2(aq) | 96 | 60.0843 | 0.001597755 |
| Al+++ | 3.5 | 26.9815 | 0.000129719 |
| Ca++ | 539 | 40.08 | 0.013448104 |
| Mg++ | 45 | 24.305 | 0.001851471 |
| Fe++ | 44 | 55.847 | 0.000787867 |
| K+ | 1910 | 39.0983 | 0.048851229 |
| Na+ | 36500 | 22.9898 | 1.587660615 |
| Sr++ | 14 | 87.62 | 0.000159781 |
| F- | 6.1 | 18.9984 | 0.00032108 |
| B(OH)3 | 412 | 61.8329 | 0.006663119 |
| Br- | 99 | 79.904 | 0.001238987 |
| Ba++ | 14 | 137.33 | 0.000101944 |
| Li+ | 91 | 6.941 | 0.013110503 |
| NH3 | 33 | 17.034 | 0.001937302 |
The MOOSE model begins by defining all the basis species — those in Table 3 as well as H2O, H+ (to fix the pH) and O2(aq) (to allow for redox couples, in particular HS- and Fe+++) — as well as the minerals of interest, which are those in Table 2 with the exception of Fe-Chlorite since it is not in the database:
[UserObjects<<<{"href": "../../../syntax/UserObjects/index.html"}>>>]
[definition]
type = GeochemicalModelDefinition<<<{"description": "User object that parses a geochemical database file, and only retains information relevant to the current geochemical model", "href": "../../../source/userobjects/GeochemicalModelDefinition.html"}>>>
database_file<<<{"description": "The name of the geochemical database file"}>>> = "../../../../geochemistry/database/moose_geochemdb.json"
basis_species<<<{"description": "A list of basis components relevant to the aqueous-equilibrium problem. H2O must appear first in this list. These components must be chosen from the 'basis species' in the database, the sorbing sites (if any) and the decoupled redox states that are in disequilibrium (if any)."}>>> = "H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq)"
equilibrium_minerals<<<{"description": "A list of minerals that are in equilibrium with the aqueous solution. All members of this list must be in the 'minerals' section of the database file"}>>> = "Siderite Pyrrhotite Dolomite Illite Anhydrite Calcite Quartz K-feldspar Kaolinite Barite Celestite Fluorite Albite Chalcedony Goethite"
[]
[]None of the input files in this page set remove_all_extrapolated_secondary_species = true in the GeochemicalModelDefinition. Setting this flag to true removes the odd secondary species whose equilibrium constants have only been measured for a small range of temperatures, since extrapolating the experimental results can lead to unrealistic values that cause convergence issues. It is simply fortunate that the simulations on this page do not need this flag set. Generally, this flag should be set true in models involving high temperatures.
A TimeIndependentReactionSolver defines:
the swaps mentioned above (so the measured concentrations of NH3 and HS- can be used instead of NO3- and O2(aq))
the charge-balance species, which is assumed to be Cl-
the bulk mole numbers of species from Table 3 and the pH (instead of mole numbers, the geochemistry module can accept other units such as mg, if more convenient)
the temperature
that no minerals are allowed to precipitate in this exploratory simulation.
[TimeIndependentReactionSolver<<<{"href": "../../../syntax/TimeIndependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = "NO3- O2(aq)"
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = " NH3 HS-"
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = "Cl-"
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = "H2O H+ Cl- SO4-- HCO3- HS- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NH3"
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = " 1.0 3.467E-7 1.619044933 0.062774835 0.065489838 0.003840583 0.001597755 0.000129719 0.013448104 0.001851471 0.000787867 0.048851229 1.587660615 0.000159781 0.00032108 0.006663119 0.001238987 0.000101944 0.013110503 0.001937302"
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = "kg_solvent_water activity bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition"
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg dimensionless moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles"
prevent_precipitation<<<{"description": "Mineral species in this list will be prevented from precipitating, irrespective of their saturation index (unless they are initially in the basis)"}>>> = "Barite Siderite Pyrrhotite Dolomite Illite Anhydrite Calcite Quartz K-feldspar Kaolinite Celestite Fluorite Albite Chalcedony Goethite"
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # not needed in this simple problem
temperature<<<{"description": "The temperature (degC) of the aqueous solution"}>>> = 25
stoichiometric_ionic_str_using_Cl_only<<<{"description": "If set to true, the stoichiometric ionic strength will be set equal to Cl- molality (or max_ionic_strength if the Cl- molality is too big). This flag overrides ionic_str_using_basis_molality_only"}>>> = true
precision<<<{"description": "Precision for printing values"}>>> = 5
[]The MOOSE output includes overall information:
Mass of solvent water = 1kg
Total mass = 1.103kg
Mass of aqueous solution = 1.103kg (without free minerals)
pH = 6.46
pe = -2.959
Ionic strength = 1.634mol/kg(solvent water)
Stoichiometric ionic strength = 1.704mol/kg(solvent water)
Activity of water = 0.9439
Temperature = 25
and reveals that many minerals are supersaturated, as expected because this water was sourced from an aquifer containing minerals and then cooled:
Minerals:
Illite = 5*H2O - 8*H+ + 3.5*SiO2(aq) + 2.3*Al+++ + 0.25*Mg++ + 0.6*K+; log10K = 9.796; SI = 9.792
Kaolinite = 5*H2O - 6*H+ + 2*SiO2(aq) + 2*Al+++; log10K = 7.434; SI = 7.731
K-feldspar = 2*H2O - 4*H+ + 3*SiO2(aq) + 1*Al+++ + 1*K+; log10K = 0.06546; SI = 7.166
Albite = 2*H2O - 4*H+ + 3*SiO2(aq) + 1*Al+++ + 1*Na+; log10K = 3.081; SI = 5.703
Pyrrhotite = -0.125*H2O - 0.7188*H+ + 0.03125*SO4-- + 0.875*Fe++ + 0.9688*HS-; log10K = -5.557; SI = 3.52
Barite = 1*SO4-- + 1*Ba++; log10K = -9.973; SI = 2.614
Quartz = 1*SiO2(aq); log10K = -4.01; SI = 1.376
Chalcedony = 1*SiO2(aq); log10K = -3.738; SI = 1.104
Siderite = -1*H+ + 1*HCO3- + 1*Fe++; log10K = -0.2225; SI = 0.8189
Dolomite = -2*H+ + 2*HCO3- + 1*Ca++ + 1*Mg++; log10K = 2.516; SI = 0.3859
Calcite = -1*H+ + 1*HCO3- + 1*Ca++; log10K = 1.711; SI = 0.01748
Fluorite = 1*Ca++ + 2*F-; log10K = -10.97; SI = -0.1147
Celestite = 1*SO4-- + 1*Sr++; log10K = -6.443; SI = -0.6262
Anhydrite = 1*SO4-- + 1*Ca++; log10K = -4.274; SI = -1.118
One other useful piece of information that is reported is the bulk composition of H+, which is 0.019675774mol.
Geochemical technicalities
At this point, it is worth recognising a couple of apparently strange features of the analyses presented in Table 1 and Table 2.
Barite is supersaturated. If it is allowed to precipitate then it will remove almost all of the Ba++ found in solution, seemingly contradicting Table 1. According to Table 2 Barite is found in only very small concentrations, so mechanisms must be present to prevent its precipitation, such as kinetic laws.
Both Chalcedony and Quartz are present in Table 2. Both are SiO2, but Quartz is more stable, so over time all Chalcedony should disappear. Kinetic rates may be responsible for these observations.
These complexities (and other more subtle ones) are completely ignored in the following, since the purpose of this presentation is to describe how to use the geochemistry module and not to get stuck on technical details of the geochemistry, even if they turn out to be important in real life.
A geochemical model of the Weber-Tensleep formation at 92C
The temperature of the Weber-Tensleep aquifer is approximately 92C, and its porosity is approximately 0.1. To build a geochemical model of the formation, the following process is used:
The water with composition described by Table 3 is equilibrated at 25C while preventing any precipitation of minerals (as in the previous section)
The pH constraint is removed (so no more H+ is allowed to enter the system)
The system is brought to 92C, allowing precipitation
The system is assumed to have volume 1L, or 1000cm, so is brought into contact with 9000cm of minerals with composition shown Table 2. The porosity is therefore close to (it is not exactly 0.1 due to the minor amounts of precipitation at step 4).
The resulting system is brought to equilibrium, allowing minerals to precipitate or dissolve if required, and assuming that no minerals are governed by kinetic laws
It is convenient to use the equilibrium model of the previous section to model steps 1 and 2. The bulk composition of H+ (0.019675774mol) is used, and the source mineral species are shown in Table 4. Chalcedony is not included because it dissolves in favor of Quartz, and Fe-Chlorite is not included because it does not appear in the database. To ensure the final porosity is close to 0.1,
Table 4: Mineralogy used in the model, where the porosity is assumed to be 0.1
| Mineral | Measured vol (%) | Model vol (%) | Model vol (cm) | Molar volume (cm.mol) | Moles |
|---|---|---|---|---|---|
| Siderite | 2 | 2 | 180 | 28.63 | 6.287111422 |
| Pyrrhotite | 0.1 | 0.1 | 9 | 17.62 | 0.510783201 |
| Dolomite | 2 | 2 | 180 | 64.365 | 2.796550921 |
| Illite | 1 | 1 | 90 | 138.94 | 0.647761624 |
| Chalcedony | 0.11 | 0 | 0 | 22.688 | 0 |
| Anhydrite | 0.6 | 0.6 | 54 | 45.94 | 1.175446234 |
| Calcite | 5 | 5 | 450 | 36.934 | 12.1838956 |
| Quartz | 80.7 | 81.299885 | 7316.98965 | 22.688 | 322.504833 |
| K-feldspar | 8 | 8 | 720 | 108.87 | 6.613392119 |
| Kaolinite | 6.60E-05 | 6.60E-05 | 0.00594 | 99.52 | 5.96865E-05 |
| Fe-Chlorite | 1 | 0 | 0 | 1 | 0 |
| Barite | 4.90E-05 | 4.90E-05 | 0.00441 | 52.1 | 8.46449E-05 |
| Goethite | 0 | 0 | 0 | 20.82 | 0 |
| Celestite | 0 | 0 | 0 | 46.25 | 0 |
| Fluorite | 0 | 0 | 0 | 24.54 | 0 |
| Albite | 0 | 0 | 0 | 100.07 | 0 |
The MOOSE input file uses a TimeDependentReactionSolver to raise the temperature and add the source minerals:
the first part of this input-file block is identical to the model above, save for fixing the bulk composition of H+ instead of fixing the pH
the second part adds the source minerals at rate given by Table 4 at a temperature of 95C so that the final temperature is around 92C as desired.
[TimeDependentReactionSolver<<<{"href": "../../../syntax/TimeDependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = "NO3- O2(aq)"
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = " NH3 HS-"
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = "Cl-"
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = "H2O H+ Cl- SO4-- HCO3- HS- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NH3"
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = " 1.0 0.019675774 1.619044933 0.062774835 0.065489838 0.003840583 0.001597755 0.000129719 0.013448104 0.001851471 0.000787867 0.048851229 1.587660615 0.000159781 0.00032108 0.006663119 0.001238987 0.000101944 0.013110503 0.001937302"
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = "kg_solvent_water bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition"
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles"
prevent_precipitation<<<{"description": "Mineral species in this list will be prevented from precipitating, irrespective of their saturation index (unless they are initially in the basis)"}>>> = "Celestite Fluorite Albite Chalcedony Goethite"
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # not needed in this simple problem
initial_temperature<<<{"description": "The initial aqueous solution is equilibrated at this system before adding reactants, changing temperature, etc."}>>> = 25
temperature<<<{"description": "Temperature. This has two different meanings if mode!=4. (1) If no species are being added to the solution (no source_species_rates are positive) then this is the temperature of the aqueous solution. (2) If species are being added, this is the temperature of the species being added. In case (2), the final aqueous-solution temperature is computed assuming the species are added, temperature is equilibrated and then, if species are also being removed, they are removed. If you wish to add species and simultaneously alter the temperature, you will have to use a sequence of heat-add-heat-add, etc steps. In the case that mode=4, temperature is the final temperature of the aqueous solution"}>>> = 95 # so final temp = 92
execute_console_output_on<<<{"description": "When to execute the geochemistry console output"}>>> = 'initial timestep_end'
source_species_names<<<{"description": "The name of the species that are added at rates given in source_species_rates. There must be an equal number of these as source_species_rates."}>>> = "Siderite Pyrrhotite Dolomite Illite Anhydrite Calcite Quartz K-feldspar Kaolinite Barite"
source_species_rates<<<{"description": "Rates, in mols/time_unit, of addition of the species with names given in source_species_names. A negative value corresponds to removing a species: be careful that you don't cause negative mass problems!"}>>> = "6.287111422 0.510783201 2.796550921 0.647761624 1.175446234 12.1838956 322.504833 6.613392119 5.96865E-05 8.46449E-05"
solver_info<<<{"description": "Print information (to the console) from the solver including residuals, swaps, etc"}>>> = true
stoichiometric_ionic_str_using_Cl_only<<<{"description": "If set to true, the stoichiometric ionic strength will be set equal to Cl- molality (or max_ionic_strength if the Cl- molality is too big). This flag overrides ionic_str_using_basis_molality_only"}>>> = true
[]The MOOSE output includes the summary:
Mass of solvent water = 0.9979kg
Total mass = 25.23kg
Mass of aqueous solution = 1.096kg (without free minerals)
pH = 6.899
pe = -3.376
Ionic strength = 1.576mol/kg(solvent water)
Stoichiometric ionic strength = 1.432mol/kg(solvent water)
Activity of water = 0.9532
Temperature = 92.06
The MOOSE output also includes the composition in the species basis that consists of the minerals and free species as shown in Table 5.
Table 5: Composition of the model at 92C expressed in the basis that includes minerals and free species. The mass of solvent water is 0.99778351kg.
| Species | Bulk composition (moles) | Free |
|---|---|---|
| Quartz | 322.4086 | 322.41188 moles |
| Calcite | 12.111108 | 12.111218 moles |
| K-feldspar | 6.8269499 | 6.8276215 moles |
| Siderite | 6.2844304 | 6.2829261 moles |
| Dolomite | 2.8670301 | 2.8672731 moles |
| Anhydrite | 1.1912027 | 1.1684061 moles |
| Pyrrhotite | 0.51474767 | 0.51549555 moles |
| Illite | 0.3732507 | 0.36893829 moles |
| Kaolinite | 0.20903322 | 0.21365601 moles |
| Barite | 0.0001865889 | 0.0001853394 moles |
| Na+ | 1.5876606 | 1.4840437 molal |
| Cl- | 1.5059455 | 1.4321212 molal |
| SO4– | 0.046792579 | 0.031819079 molal |
| Li+ | 0.013110503 | 0.012928063 molal |
| B(OH)3 | 0.006663119 | 0.0014134967 molal |
| Br- | 0.001238987 | 0.0012417393 molal |
| F- | 0.00032108 | 0.00023740743 molal |
| Sr++ | 0.000159781 | 0.00013115377 molal |
| NH3 | 0.001937302 | 3.1632078e-07 molal |
It is interesting to compare the concentration of species in the model when the aqueous solution is removed from the mineral assemblage. This information is also outputted by the geochemistry module and is shown in Table 6:
The concentration of Cl- is impacted by charge-neutrality
The concentration of HCO3-, Al+++, K+ and Ba++ are all much less in the model compared with the observation due to mineral precipitation
The concentration of species not impacted by minerals is identical (Na+, Sr++, F-, B, Br-, Li+, NH3)
The model's concentration of the remaining species are similar to the observations
Table 6: Composition at 92C after removing minerals in comparison with the original measurements.
| Species | Measured conc (molal) | Model conc (molal) |
|---|---|---|
| Cl- | 1.619044933 | 1.5059455 |
| SO4– | 0.062774835 | 0.068842508 |
| HCO3- | 0.065489838 | 0.00090808324 |
| HS- | 0.003840583 | 0.0029683017 |
| SiO2(aq) | 0.001597755 | 0.00055318842 |
| Al+++ | 0.000129719 | 1.3497527e-06 |
| Ca++ | 0.013448104 | 0.022443348 |
| Mg++ | 0.001851471 | 0.00083515093 |
| Fe++ | 0.000787867 | 0.00084984977 |
| K+ | 0.048851229 | 0.0019158317 |
| Na+ | 1.587660615 | 1.5876606 |
| Sr++ | 0.000159781 | 0.000159781 |
| F- | 0.00032108 | 0.00032108 |
| B(OH)3 | 0.006663119 | 0.006663119 |
| Br- | 0.001238987 | 0.001238987 |
| Ba++ | 0.000101944 | 1.2495026e-06 |
| Li+ | 0.013110503 | 0.013110503 |
| NH3 | 0.001937302 | 0.001937302 |
The modelling also reveals that Goethite, Albite and Flourite are all supersaturated, so will probably precipitate if they are in equilibrium with the aqueous solution, apparently contradicting Table 2.
Goethite = 3.255*H2O - 0.129*Pyrrhotite - 3.345*Kaolinite + 0.8548*Calcite + 0.129*Anhydrite - 0.9839*Dolomite - 2.361*K-feldspar + 3.935*Illite + 1.113*Siderite; log10K = -1.607; SI = 1.539
Albite = 0.4*H2O + 0.5*SO4-- - 1.2*Kaolinite + 2*Quartz + 1*Calcite - 0.5*Anhydrite - 0.5*Dolomite - 1.2*K-feldspar + 2*Illite + 1*Na+; log10K = -2.122; SI = 0.8307
Fluorite = -1*SO4-- + 1*Anhydrite + 2*F-; log10K = -5.468; SI = 0.2909
Chalcedony = 1*Quartz; log10K = 0.2214; SI = -0.2214
Celestite = 1*SO4-- + 1*Sr++; log10K = -6.861; SI = -0.4025
The total mineral volume is reported to be 9005.244392233cm. To ensure the porosity is exactly 0.1, the free amount of Quartz is reduced by cmmol in the reactive transport simulations, leading to a bulk composition of mol.
This completes the definition of the geochemical system used in this presentation. In summary:
the bulk composition is determined by Table 5 in the basis of that table, amending Quartz to have a bulk composition of mol (the geochemistry module can use other units such as g, mg, etc, but this page uses mole-based units exclusively);
all minerals in the basis of Table 5 are assumed to be at equilibrium with the aqueous system (the
geochemistrymodule can easily handle kinetic reactions — a simple GeoTES example may be found here — but no kinetics are used in this presentation);the minerals Goethite, Albite, and Flourite are assumed to never precipitate
Scaling and heating the aqueous solution to 160C
The geochemical system just defined may be heated to 160C in order to explore potential scaling in a heat exchanger. The following method is used:
The aqueous solution is removed from the aquifer, by removing all free minerals are removed from the model just defined
The aqueous solution is slowly heated from 92C to 160C, immediately removing any precipitates as soon as they form
This technique is studying the "worst case" scenario for scaling in a heat exchanger, for it is possible that a precipitate forms only to dissolve upon further heating, but the technique used removes the precipitate and prevents its dissolution.
The TimeDependentReactionSolver defines:
the swaps needed to bring the minerals into the basis, so it is as described in Table 5;
the bulk composition of the resulting basis (from Table 5, amending Quartz to have a bulk composition of mol, recall that the geochemistry module can use other units such as grams or mg if convenient, but this page uses mole-based units exclusively);
that the Fluorite, Albite and Goethite minerals will not precipitate;
the initial temperature;
that
mode = 1is used, which means all precipitates are removed at the start of each time step;the temperature is controlled by the AuxVariable called
temp_controller.
[TimeDependentReactionSolver<<<{"href": "../../../syntax/TimeDependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = "NO3- H+ Fe++ Ba++ SiO2(aq) Mg++ O2(aq) Al+++ K+ Ca++ HCO3-"
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = " NH3 Pyrrhotite K-feldspar Barite Quartz Dolomite Siderite Calcite Illite Anhydrite Kaolinite"
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = "Cl-"
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = "H2O Quartz Calcite K-feldspar Siderite Dolomite Anhydrite Pyrrhotite Illite Kaolinite Barite Na+ Cl- SO4-- Li+ B(OH)3 Br- F- Sr++ NH3"
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = " 0.99778351 322.177447 12.111108 6.8269499 6.2844304 2.8670301 1.1912027 0.51474767 0.3732507 0.20903322 0.0001865889 1.5876606 1.5059455 0.046792579 0.013110503 0.006663119 0.001238987 0.00032108 0.000159781 0.001937302"
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = "kg_solvent_water bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition"
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles"
prevent_precipitation<<<{"description": "Mineral species in this list will be prevented from precipitating, irrespective of their saturation index (unless they are initially in the basis)"}>>> = "Fluorite Albite Goethite"
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # not needed in this simple problem
initial_temperature<<<{"description": "The initial aqueous solution is equilibrated at this system before adding reactants, changing temperature, etc."}>>> = 92
mode<<<{"description": "This may vary temporally. If mode=1 then 'dump' mode is used, which means all non-kinetic mineral masses are removed from the system before the equilibrium solution is sought (ie, removal occurs at the beginning of the time step). If mode=2 then 'flow-through' mode is used, which means all mineral masses are removed from the system after it the equilbrium solution has been found (ie, at the end of a time step). If mode=3 then 'flush' mode is used, then before the equilibrium solution is sought (ie, at the start of a time step) water+species is removed from the system at the same rate as pure water + non-mineral solutes are entering the system (specified in source_species_rates). If mode=4 then 'heat-exchanger' mode is used, which means the entire current aqueous solution is removed, then the source_species are added, then the temperature is set to 'cold_temperature', the system is solved and any precipitated minerals are removed, then the temperature is set to 'temperature', the system re-solved and any precipitated minerals are removed. If mode is any other number, no special mode is active (the system simply responds to the source_species_rates, controlled_activity_value, etc)."}>>> = 1 # dump all minerals at the start of each time-step
temperature<<<{"description": "Temperature. This has two different meanings if mode!=4. (1) If no species are being added to the solution (no source_species_rates are positive) then this is the temperature of the aqueous solution. (2) If species are being added, this is the temperature of the species being added. In case (2), the final aqueous-solution temperature is computed assuming the species are added, temperature is equilibrated and then, if species are also being removed, they are removed. If you wish to add species and simultaneously alter the temperature, you will have to use a sequence of heat-add-heat-add, etc steps. In the case that mode=4, temperature is the final temperature of the aqueous solution"}>>> = temp_controller
execute_console_output_on<<<{"description": "When to execute the geochemistry console output"}>>> = '' # only CSV output for this problem
stoichiometric_ionic_str_using_Cl_only<<<{"description": "If set to true, the stoichiometric ionic strength will be set equal to Cl- molality (or max_ionic_strength if the Cl- molality is too big). This flag overrides ionic_str_using_basis_molality_only"}>>> = true
[]The Executioner gives meaning to time:
[Executioner<<<{"href": "../../../syntax/Executioner/index.html"}>>>]
type = Transient
dt = 0.01
end_time = 1.0
[]The temp_controller is a simple FunctionAux:
[temp_controller_auxk]
type = FunctionAux
variable = temp_controller
function = '92 + (160 - 92) * t'
execute_on = timestep_begin
[]and the moles dumped to the heat exchanger (scaling) are recorded by a sequence of GeochemistryQuantityAux:
[Anhydrite_mol_auxk]
type = GeochemistryQuantityAux
reactor = reactor
variable = Anhydrite_mol
species = Anhydrite
quantity = moles_dumped
[]Recording the results into Postprocessors yields the results shown in Figure 2. It is interesting to compare this with Nic Spycher's model of the Weber-Tensleep formation which predicts:
around 2kg/m(formation water) of Anhydrite, or around 0.7cm/L(formation water), which is quite similar to the current model;
around 0.03cm/L(formation water) of Dolomite, 0.01cm/L(formation water) of Calcite, 0.0025cm/L(formation water) of Siderite, while the current model predicts none of these. This could be due to a combination of a different activity model, a different geochemical model (the pH is lower), and a different definition of "scaling".
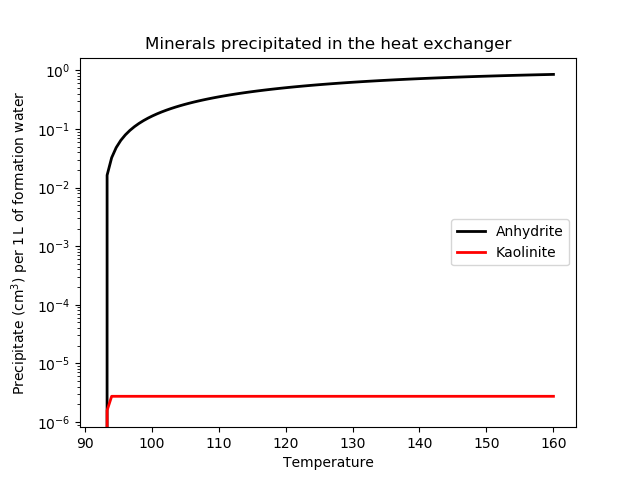
Figure 2: Degree of scaling precipitate expected in the heat exchanger according to the model of formation water.
Spatial aquifer geochemistry input file
Having created the geochemical model at 92C, it is easy to create a version that includes spatial dependence. Although the input file may be run by itself, no interesting phenomena will be observed since the temperature will be fixed and source-species rates will be zero. When coupled with a porous_flow input file (described below) the temperature and source-species rates will be non-trivial. The input file begins with a GeochemicalModelDefinition:
[UserObjects<<<{"href": "../../../syntax/UserObjects/index.html"}>>>]
[definition]
type = GeochemicalModelDefinition<<<{"description": "User object that parses a geochemical database file, and only retains information relevant to the current geochemical model", "href": "../../../source/userobjects/GeochemicalModelDefinition.html"}>>>
database_file<<<{"description": "The name of the geochemical database file"}>>> = '../../../../geochemistry/database/moose_geochemdb.json'
basis_species<<<{"description": "A list of basis components relevant to the aqueous-equilibrium problem. H2O must appear first in this list. These components must be chosen from the 'basis species' in the database, the sorbing sites (if any) and the decoupled redox states that are in disequilibrium (if any)."}>>> = 'H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq)'
equilibrium_minerals<<<{"description": "A list of minerals that are in equilibrium with the aqueous solution. All members of this list must be in the 'minerals' section of the database file"}>>> = 'Siderite Pyrrhotite Dolomite Illite Anhydrite Calcite Quartz K-feldspar Kaolinite Barite Celestite Fluorite Albite Chalcedony Goethite'
[]
[nodal_void_volume_uo]
type = NodalVoidVolume<<<{"description": "UserObject to compute the nodal void volume. Take care if you block-restrict this UserObject, since the volumes of the nodes on the block's boundary will not include any contributions from outside the block.", "href": "../../../source/userobjects/NodalVoidVolume.html"}>>>
porosity<<<{"description": "Porosity"}>>> = porosity
execute_on<<<{"description": "The list of flag(s) indicating when this object should be executed. For a description of each flag, see https://mooseframework.inl.gov/source/interfaces/SetupInterface.html."}>>> = 'initial timestep_end' # initial means this is evaluated properly for the first timestep
[]
[]The SpatialReactionSolver defines:
the swaps needed to bring the basis to the state defined in Table 5
the initial composition of Table 5 (amending Quartz to have a bulk composition of mol). Please note the key concept that each finite element node considers just 1 litre of aqueous solution
the
initial_temperaturethat the temperature at each finite-element node will be controlled by the
temperatureAuxVariable (which will be provided by the parentporous_flowsimulation)the source species, and their rates per 1 litre of aqueous solution
that various AuxVariables are not needed in this simulation
[SpatialReactionSolver<<<{"href": "../../../syntax/SpatialReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = 'Cl-'
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = 'NO3- H+ Fe++ Ba++ SiO2(aq) Mg++ O2(aq) Al+++ K+ Ca++ HCO3-'
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = ' NH3 Pyrrhotite K-feldspar Barite Quartz Dolomite Siderite Calcite Illite Anhydrite Kaolinite'
# ASSUME that 1 litre of solution contains:
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = 'H2O Quartz Calcite K-feldspar Siderite Dolomite Anhydrite Pyrrhotite Illite Kaolinite Barite Na+ Cl- SO4-- Li+ B(OH)3 Br- F- Sr++ NH3'
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = ' 0.99778351 322.177447 12.111108 6.8269499 6.2844304 2.8670301 1.1912027 0.51474767 0.3732507 0.20903322 0.0001865889 1.5876606 1.5059455 0.046792579 0.013110503 0.006663119 0.001238987 0.00032108 0.000159781 0.001937302'
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = 'kg_solvent_water bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition bulk_composition'
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles moles"
prevent_precipitation<<<{"description": "Mineral species in this list will be prevented from precipitating, irrespective of their saturation index (unless they are initially in the basis)"}>>> = 'Fluorite Albite Goethite'
initial_temperature<<<{"description": "Temperature at which the initial system is equilibrated. This is uniform over the entire mesh."}>>> = 92
temperature<<<{"description": "Temperature"}>>> = temperature
source_species_names<<<{"description": "The name of the species that are added at rates given in source_species_rates. There must be an equal number of these as source_species_rates."}>>> = 'H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq) H2O'
source_species_rates<<<{"description": "Rates, in mols/time_unit, of addition of the species with names given in source_species_names. A negative value corresponds to removing a species: be careful that you don't cause negative mass problems!"}>>> = ' rate_H_per_1l rate_Cl_per_1l rate_SO4_per_1l rate_HCO3_per_1l rate_SiO2aq_per_1l rate_Al_per_1l rate_Ca_per_1l rate_Mg_per_1l rate_Fe_per_1l rate_K_per_1l rate_Na_per_1l rate_Sr_per_1l rate_F_per_1l rate_BOH_per_1l rate_Br_per_1l rate_Ba_per_1l rate_Li_per_1l rate_NO3_per_1l rate_O2aq_per_1l rate_H2O_per_1l'
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # max_ionic_strength in such a simple problem does not need ramping
execute_console_output_on<<<{"description": "When to execute the geochemistry console output"}>>> = '' # only CSV and exodus output for this simulation
add_aux_molal<<<{"description": "Add AuxVariables measured in molal units (ie mol(species)/kg(solvent_water)). These are named molal_name, where 'name' is the species name. AuxVariables are added for all species except minerals"}>>> = false # save some memory and reduce variables in output exodus
add_aux_mg_per_kg<<<{"description": "Add AuxVariables measured in mg(species)/kg(solvent_water). These are named mg_per_kg_name, where 'name' is the species name. AuxVariables are added for all species except minerals"}>>> = false # save some memory and reduce variables in output exodus
add_aux_free_mg<<<{"description": "Add AuxVariables for all minerals measured in free mg. These are named free_mg_name, where 'name' is the species name"}>>> = false # save some memory and reduce variables in output exodus
add_aux_activity<<<{"description": "Add AuxVariables that record the activity for all species (for gas species this equals the gas fugacity). These are called activity_name where 'name' is the species name."}>>> = false # save some memory and reduce variables in output exodus
add_aux_bulk_moles<<<{"description": "Add AuxVariables that record the number of bulk-composition moles for all species. Note that these will be zero for any species not currently in the basis. These are called bulk_moles_name where 'name' is the species name."}>>> = false # save some memory and reduce variables in output exodus
adaptive_timestepping<<<{"description": "Use adaptive timestepping at each node in an attempt to ensure convergence of the solver. Setting this parameter to false saves some compute time because copying of datastructures is avoided"}>>> = true
[]The remainder of the input file is mostly concerned with translating between porous-flow information (mass fractions and source rates at each node) and geochemistry information (moles in each litre of fluid). This block translates the porous_flow rate, which is the rate of change of a species mass at each node (in kg.s) into a rate of change per 1 litre of aqueous solution at each node:
[rate_H_per_1l_auxk]
type = ParsedAux
coupled_variables = 'pf_rate_H nodal_void_volume'
variable = rate_H_per_1l
expression = 'pf_rate_H / 1.0079 / nodal_void_volume'
execute_on = 'timestep_end'
[]Here 1.0079 is the molar mass (g.mol) of the species H, and nodal_void_volume is the volume of aqueous solution held at each node. This block translates the geochemistry moles of transported H into a mass fraction of H:
[massfrac_H_auxk]
type = ParsedAux
coupled_variables = 'transported_H transported_mass'
variable = massfrac_H
expression = 'transported_H * 1.0079 / transported_mass'
execute_on = 'timestep_end'
[]using the transported_mass, which is
[transported_mass_auxk]
type = ParsedAux
coupled_variables = ' transported_H transported_Cl transported_SO4 transported_HCO3 transported_SiO2aq transported_Al transported_Ca transported_Mg transported_Fe transported_K transported_Na transported_Sr transported_F transported_BOH transported_Br transported_Ba transported_Li transported_NO3 transported_O2aq transported_H2O'
variable = transported_mass
expression = 'transported_H * 1.0079 + transported_Cl * 35.453 + transported_SO4 * 96.0576 + transported_HCO3 * 61.0171 + transported_SiO2aq * 60.0843 + transported_Al * 26.9815 + transported_Ca * 40.08 + transported_Mg * 24.305 + transported_Fe * 55.847 + transported_K * 39.0983 + transported_Na * 22.9898 + transported_Sr * 87.62 + transported_F * 18.9984 + transported_BOH * 61.8329 + transported_Br * 79.904 + transported_Ba * 137.33 + transported_Li * 6.941 + transported_NO3 * 62.0049 + transported_O2aq * 31.9988 + transported_H2O * 18.01801802'
execute_on = 'timestep_end'
[]The porosity is calculated using the free mineral volumes:
[porosity_auxk]
type = ParsedAux
coupled_variables = 'free_cm3_Siderite free_cm3_Pyrrhotite free_cm3_Dolomite free_cm3_Illite free_cm3_Anhydrite free_cm3_Calcite free_cm3_Quartz free_cm3_Kfeldspar free_cm3_Kaolinite free_cm3_Barite free_cm3_Celestite free_cm3_Fluorite free_cm3_Albite free_cm3_Chalcedony free_cm3_Goethite'
expression = '1000.0 / (1000.0 + free_cm3_Siderite + free_cm3_Pyrrhotite + free_cm3_Dolomite + free_cm3_Illite + free_cm3_Anhydrite + free_cm3_Calcite + free_cm3_Quartz + free_cm3_Kfeldspar + free_cm3_Kaolinite + free_cm3_Barite + free_cm3_Celestite + free_cm3_Fluorite + free_cm3_Albite + free_cm3_Chalcedony + free_cm3_Goethite)'
variable = porosity
execute_on = 'timestep_end'
[]Injection, production and porous flow
The Weber-Tensleep aquifer is around 200m thick, and injecting and producing over its entire thickness may lead to unacceptably low efficiencies as the buoyant hot water rises to the top of the aquifer, never to be recovered. For the purposes of this example, assume there is a 10m thick horizontal aquifer, bounded above and below by caps. The physical properties are listed in Table 7.
Table 7: Physical properties of the aquifer and caps
| Property | Quantity |
|---|---|
| Aquifer thickness | 10m |
| Aquifer depth | 3000m |
| Aquifer initial porepressure | 30MPa |
| Aquifer initial temperature | 92C |
| Aquifer horizontal permeability | m |
| Aquifer vertical permeability | m |
| Aquifer porosity | 0.1 |
| Aquifer thermal conductivity | 1.3W.m.K |
| Cap thickness | 20m |
| Cap isotropic permeability | m |
| Cap porosity | 0.01 |
| Cap thermal conductivity | 1.3W.m.K |
| Geothermal gradient | 0 |
As shown in Figure 1, the well geometry consists of a single vertical well injecting over the entire aquifer thickness and a single vertical well producing over the entire aquifer thickness. Only the injection phase of the GeoTES system is explored here, with parameters listed in Table 8. The injection and production rates are chosen so that the porepressure remains positive at the production well given the relatively low aquifer permeability.
Table 8: Injection parameters of the GeoTES system
| Property | Quantity |
|---|---|
| Injection and production rate | 0.2kg.s |
| Injection temperature | 160C |
| Injection fluid | Heated production water: all precipitates removed |
| Injection time | 90days |
| Distance between wells | 50m |
The injection-production simulation may be performed using the porous_flow module. Because this presentation is focussing on geochemistry, only the highlights of the porous_flow input file are mentioned.
The variables are the mass-fractions of each transported species (which are those in the original geochemical basis before any swaps), called f_H, f_Cl, f_SO4, etc, with initial condition defined by Table 6 and converted into mass-fractions as shown in Table 9.
Table 9: Composition at 92C of transported species (ie, not including minerals) expressed in the original basis.
| Species | Moles | Molar weight (g.mol) | Mass (g) | Mass fraction |
|---|---|---|---|---|
| H2O | 55.38 | 18.01801802 | 997.8378378 | 0.910314278 |
| H+ | -0.003232 | 1.0079 | -0.003257533 | -2.9718E-06 |
| Cl- | 1.506 | 35.453 | 53.392218 | 0.048709015 |
| SO4– | 0.06887 | 96.0576 | 6.615486912 | 0.006035221 |
| HCO3- | 0.0009061 | 61.0171 | 0.055287594 | 5.04381E-05 |
| SiO2(aq) | 0.0005525 | 60.0843 | 0.033196576 | 3.02848E-05 |
| Al+++ | 1.35E-06 | 26.9815 | 3.6479E-05 | 3.32793E-08 |
| Ca++ | 0.02247 | 40.08 | 0.9005976 | 0.000821603 |
| Mg++ | 0.0008366 | 24.305 | 0.020333563 | 1.855E-05 |
| Fe++ | 0.0008498 | 55.847 | 0.047458781 | 4.3296E-05 |
| K+ | 0.001913 | 39.0983 | 0.074795048 | 6.82345E-05 |
| Na+ | 1.588 | 22.9898 | 36.5078024 | 0.033305586 |
| Sr++ | 0.0001598 | 87.62 | 0.014001676 | 1.27735E-05 |
| F- | 0.0003211 | 18.9984 | 0.006100386 | 5.5653E-06 |
| B(OH)3 | 0.006663 | 61.8329 | 0.411992613 | 0.000375855 |
| Br- | 0.001239 | 79.904 | 0.099001056 | 9.03174E-05 |
| Ba++ | 1.25E-06 | 137.33 | 0.000171388 | 1.56355E-07 |
| Li+ | 0.01311 | 6.941 | 0.09099651 | 8.30149E-05 |
| NO3- | 0.001937 | 62.0049 | 0.120103491 | 0.000109569 |
| O2(aq) | -0.002426 | 31.9988 | -0.077629089 | -7.082E-05 |
To interface with the child geochemistry simulation (detailed above), the rates of change of each transported species must be recorded. This is performed via the save_component_rate_in feature of the PorousFlowFullySaturated Action:
#########################################
# #
# File written by create_input_files.py #
# #
#########################################
# PorousFlow simulation of injection and production in a simplified GeoTES aquifer
# Much of this file is standard porous-flow stuff. The unusual aspects are:
# - transfer of the rates of changes of each species (kg.s) to the aquifer_geochemistry.i simulation. This is achieved by saving these changes from the PorousFlowMassTimeDerivative residuals
# - transfer of the temperature field to the aquifer_geochemistry.i simulation
# Interesting behaviour can be simulated by this file without its 'parent' simulation, exchanger.i. exchanger.i provides mass-fractions injected via the injection_rate_massfrac_* variables, but since these are more-or-less constant throughout the duration of the exchanger.i simulation, the initial_conditions specified below may be used. Similar, exchanger.i provides injection_temperature, but that is also constant.
injection_rate = -0.02 # kg/s/m, negative because injection as a source
production_rate = 0.02 # kg/s/m, this is about the maximum that can be sustained by the aquifer, with its fairly low permeability, without porepressure becoming negative
[Mesh<<<{"href": "../../../syntax/Mesh/index.html"}>>>]
[gen]
type = GeneratedMeshGenerator<<<{"description": "Create a line, square, or cube mesh with uniformly spaced or biased elements.", "href": "../../../source/meshgenerators/GeneratedMeshGenerator.html"}>>>
dim<<<{"description": "The dimension of the mesh to be generated"}>>> = 3
xmin<<<{"description": "Lower X Coordinate of the generated mesh"}>>> = -75
xmax<<<{"description": "Upper X Coordinate of the generated mesh"}>>> = 75
ymin<<<{"description": "Lower Y Coordinate of the generated mesh"}>>> = 0
ymax<<<{"description": "Upper Y Coordinate of the generated mesh"}>>> = 40
zmin<<<{"description": "Lower Z Coordinate of the generated mesh"}>>> = -25
zmax<<<{"description": "Upper Z Coordinate of the generated mesh"}>>> = 25
nx<<<{"description": "Number of elements in the X direction"}>>> = 15
ny<<<{"description": "Number of elements in the Y direction"}>>> = 4
nz<<<{"description": "Number of elements in the Z direction"}>>> = 5
[]
[aquifer]
type = ParsedSubdomainMeshGenerator<<<{"description": "Uses a parsed expression (`combinatorial_geometry`) to determine if an element (via its centroid) is inside the region defined by the expression and assigns a new block ID.", "href": "../../../source/meshgenerators/ParsedSubdomainMeshGenerator.html"}>>>
input<<<{"description": "The mesh we want to modify"}>>> = gen
block_id<<<{"description": "Subdomain id to set for inside of the combinatorial"}>>> = 1
block_name<<<{"description": "Subdomain name to set for inside of the combinatorial"}>>> = aquifer
combinatorial_geometry<<<{"description": "Function expression encoding a combinatorial geometry"}>>> = 'z >= -5 & z <= 5'
[]
[injection_nodes]
input<<<{"description": "The mesh we want to modify"}>>> = aquifer
type = ExtraNodesetGenerator<<<{"description": "Creates a new node set and a new boundary made with the nodes the user provides.", "href": "../../../source/meshgenerators/ExtraNodesetGenerator.html"}>>>
new_boundary<<<{"description": "The names of the boundaries to create"}>>> = injection_nodes
coord<<<{"description": "The nodes with coordinates you want to be in the nodeset. Separate multple coords with ';' (Either this parameter or \"nodes\" must be supplied)."}>>> = '-25 0 -5; -25 0 5'
[]
[production_nodes]
input<<<{"description": "The mesh we want to modify"}>>> = injection_nodes
type = ExtraNodesetGenerator<<<{"description": "Creates a new node set and a new boundary made with the nodes the user provides.", "href": "../../../source/meshgenerators/ExtraNodesetGenerator.html"}>>>
new_boundary<<<{"description": "The names of the boundaries to create"}>>> = production_nodes
coord<<<{"description": "The nodes with coordinates you want to be in the nodeset. Separate multple coords with ';' (Either this parameter or \"nodes\" must be supplied)."}>>> = '25 0 -5; 25 0 5'
[]
[]
[GlobalParams<<<{"href": "../../../syntax/GlobalParams/index.html"}>>>]
PorousFlowDictator = dictator
gravity = '0 0 -10'
[]
[BCs<<<{"href": "../../../syntax/BCs/index.html"}>>>]
[injection_temperature]
type = MatchedValueBC<<<{"description": "Implements a NodalBC which equates two different Variables' values on a specified boundary.", "href": "../../../source/bcs/MatchedValueBC.html"}>>>
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = temperature
v<<<{"description": "The variable whose value we are to match."}>>> = injection_temperature
boundary<<<{"description": "The list of boundary IDs from the mesh where this object applies"}>>> = injection_nodes
[]
[]
[FluidProperties<<<{"href": "../../../syntax/FluidProperties/index.html"}>>>]
[the_simple_fluid]
type = SimpleFluidProperties<<<{"description": "Fluid properties for a simple fluid with a constant bulk density", "href": "../../../source/fluidproperties/SimpleFluidProperties.html"}>>>
thermal_expansion<<<{"description": "Constant coefficient of thermal expansion (1/K)"}>>> = 0
bulk_modulus<<<{"description": "Constant bulk modulus (Pa)"}>>> = 2E9
viscosity<<<{"description": "Constant dynamic viscosity (Pa.s)"}>>> = 1E-3
density0<<<{"description": "Density at zero pressure and zero temperature"}>>> = 1000
cv<<<{"description": "Constant specific heat capacity at constant volume (J/kg/K)"}>>> = 4000.0
cp<<<{"description": "Constant specific heat capacity at constant pressure (J/kg/K)"}>>> = 4000.0
[]
[]
[PorousFlowFullySaturated<<<{"href": "../../../syntax/PorousFlowFullySaturated/index.html"}>>>]
coupling_type<<<{"description": "The type of simulation. For simulations involving Mechanical deformations, you will need to supply the correct Biot coefficient. For simulations involving Thermal flows, you will need an associated ConstantThermalExpansionCoefficient Material"}>>> = ThermoHydro
porepressure<<<{"description": "The name of the porepressure variable"}>>> = porepressure
temperature<<<{"description": "For isothermal simulations, this is the temperature at which fluid properties (and stress-free strains) are evaluated at. Otherwise, this is the name of the temperature variable. Units = Kelvin"}>>> = temperature
mass_fraction_vars<<<{"description": "List of variables that represent the mass fractions. With only one fluid component, this may be left empty. With N fluid components, the format is 'f_0 f_1 f_2 ... f_(N-1)'. That is, the N^th component need not be specified because f_N = 1 - (f_0 + f_1 + ... + f_(N-1)). It is best numerically to choose the N-1 mass fraction variables so that they represent the fluid components with small concentrations. This Action will associated the i^th mass fraction variable to the equation for the i^th fluid component, and the pressure variable to the N^th fluid component."}>>> = 'f_H f_Cl f_SO4 f_HCO3 f_SiO2aq f_Al f_Ca f_Mg f_Fe f_K f_Na f_Sr f_F f_BOH f_Br f_Ba f_Li f_NO3 f_O2aq '
save_component_rate_in<<<{"description": "List of AuxVariables into which the rate-of-change of each fluid component at each node will be saved. There must be exactly N of these to match the N fluid components. The result will be measured in kg/s, where the kg is the mass of the fluid component at the node (or m^3/s if multiply_by_density=false). Note that this saves the result from the MassTimeDerivative Kernels, but NOT from the MassVolumetricExpansion Kernels."}>>> = 'rate_H rate_Cl rate_SO4 rate_HCO3 rate_SiO2aq rate_Al rate_Ca rate_Mg rate_Fe rate_K rate_Na rate_Sr rate_F rate_BOH rate_Br rate_Ba rate_Li rate_NO3 rate_O2aq rate_H2O' # change in kg at every node / dt
fp<<<{"description": "The name of the user object for fluid properties. Only needed if fluid_properties_type = PorousFlowSingleComponentFluid"}>>> = the_simple_fluid
temperature_unit<<<{"description": "The unit of the temperature variable"}>>> = Celsius
[]
[Materials<<<{"href": "../../../syntax/Materials/index.html"}>>>]
[porosity_caps]
type = PorousFlowPorosityConst<<<{"description": "This Material calculates the porosity assuming it is constant", "href": "../../../source/materials/PorousFlowPorosityConst.html"}>>> # this simulation has no porosity changes from dissolution
block<<<{"description": "The list of blocks (ids or names) that this object will be applied"}>>> = 0
porosity<<<{"description": "The porosity (assumed indepenent of porepressure, temperature, strain, etc, for this material). This should be a real number, or a constant monomial variable (not a linear lagrange or other kind of variable)."}>>> = 0.01
[]
[porosity_aquifer]
type = PorousFlowPorosityConst<<<{"description": "This Material calculates the porosity assuming it is constant", "href": "../../../source/materials/PorousFlowPorosityConst.html"}>>> # this simulation has no porosity changes from dissolution
block<<<{"description": "The list of blocks (ids or names) that this object will be applied"}>>> = aquifer
porosity<<<{"description": "The porosity (assumed indepenent of porepressure, temperature, strain, etc, for this material). This should be a real number, or a constant monomial variable (not a linear lagrange or other kind of variable)."}>>> = 0.063
[]
[permeability_caps]
type = PorousFlowPermeabilityConst<<<{"description": "This Material calculates the permeability tensor assuming it is constant", "href": "../../../source/materials/PorousFlowPermeabilityConst.html"}>>>
block<<<{"description": "The list of blocks (ids or names) that this object will be applied"}>>> = 0
permeability<<<{"description": "The permeability tensor (usually in m^2), which is assumed constant for this material"}>>> = '1E-18 0 0 0 1E-18 0 0 0 1E-18'
[]
[permeability_aquifer]
type = PorousFlowPermeabilityConst<<<{"description": "This Material calculates the permeability tensor assuming it is constant", "href": "../../../source/materials/PorousFlowPermeabilityConst.html"}>>>
block<<<{"description": "The list of blocks (ids or names) that this object will be applied"}>>> = aquifer
permeability<<<{"description": "The permeability tensor (usually in m^2), which is assumed constant for this material"}>>> = '1.7E-15 0 0 0 1.7E-15 0 0 0 4.1E-16'
[]
[thermal_conductivity]
type = PorousFlowThermalConductivityIdeal<<<{"description": "This Material calculates rock-fluid combined thermal conductivity by using a weighted sum. Thermal conductivity = dry_thermal_conductivity + S^exponent * (wet_thermal_conductivity - dry_thermal_conductivity), where S is the aqueous saturation", "href": "../../../source/materials/PorousFlowThermalConductivityIdeal.html"}>>>
dry_thermal_conductivity<<<{"description": "The thermal conductivity of the rock matrix when the aqueous saturation is zero"}>>> = '0 0 0 0 0 0 0 0 0'
[]
[rock_heat]
type = PorousFlowMatrixInternalEnergy<<<{"description": "This Material calculates the internal energy of solid rock grains, which is specific_heat_capacity * density * temperature. Kernels multiply this by (1 - porosity) to find the energy density of the porous rock in a rock-fluid system", "href": "../../../source/materials/PorousFlowMatrixInternalEnergy.html"}>>>
density<<<{"description": "Density of the rock grains"}>>> = 2500.0
specific_heat_capacity<<<{"description": "Specific heat capacity of the rock grains (J/kg/K)."}>>> = 1200.0
[]
[]
[Preconditioning<<<{"href": "../../../syntax/Preconditioning/index.html"}>>>]
active<<<{"description": "If specified only the blocks named will be visited and made active"}>>> = typically_efficient
[typically_efficient]
type = SMP<<<{"description": "Single matrix preconditioner (SMP) builds a preconditioner using user defined off-diagonal parts of the Jacobian.", "href": "../../../source/preconditioners/SingleMatrixPreconditioner.html"}>>>
full<<<{"description": "Set to true if you want the full set of couplings between variables simply for convenience so you don't have to set every off_diag_row and off_diag_column combination."}>>> = true
petsc_options_iname<<<{"description": "Names of PETSc name/value pairs"}>>> = '-pc_type -pc_hypre_type'
petsc_options_value<<<{"description": "Values of PETSc name/value pairs (must correspond with \"petsc_options_iname\""}>>> = ' hypre boomeramg'
[]
[strong]
type = SMP<<<{"description": "Single matrix preconditioner (SMP) builds a preconditioner using user defined off-diagonal parts of the Jacobian.", "href": "../../../source/preconditioners/SingleMatrixPreconditioner.html"}>>>
full<<<{"description": "Set to true if you want the full set of couplings between variables simply for convenience so you don't have to set every off_diag_row and off_diag_column combination."}>>> = true
petsc_options<<<{"description": "Singleton PETSc options"}>>> = '-ksp_diagonal_scale -ksp_diagonal_scale_fix'
petsc_options_iname<<<{"description": "Names of PETSc name/value pairs"}>>> = '-pc_type -sub_pc_type -sub_pc_factor_shift_type -pc_asm_overlap'
petsc_options_value<<<{"description": "Values of PETSc name/value pairs (must correspond with \"petsc_options_iname\""}>>> = ' asm ilu NONZERO 2'
[]
[probably_too_strong]
type = SMP<<<{"description": "Single matrix preconditioner (SMP) builds a preconditioner using user defined off-diagonal parts of the Jacobian.", "href": "../../../source/preconditioners/SingleMatrixPreconditioner.html"}>>>
full<<<{"description": "Set to true if you want the full set of couplings between variables simply for convenience so you don't have to set every off_diag_row and off_diag_column combination."}>>> = true
petsc_options_iname<<<{"description": "Names of PETSc name/value pairs"}>>> = '-pc_type -pc_factor_mat_solver_package'
petsc_options_value<<<{"description": "Values of PETSc name/value pairs (must correspond with \"petsc_options_iname\""}>>> = ' lu mumps'
[]
[]
[Executioner<<<{"href": "../../../syntax/Executioner/index.html"}>>>]
type = Transient
solve_type = Newton
end_time = 7.76E6 # 90 days
[TimeStepper<<<{"href": "../../../syntax/Executioner/TimeStepper/index.html"}>>>]
type = FunctionDT
function = 'min(3E4, max(1E4, 0.2 * t))'
[]
[]
[Outputs<<<{"href": "../../../syntax/Outputs/index.html"}>>>]
exodus<<<{"description": "Output the results using the default settings for Exodus output."}>>> = true
[]
[Variables<<<{"href": "../../../syntax/Variables/index.html"}>>>]
[f_H]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = -2.952985071156e-06
[]
[f_Cl]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.04870664551708
[]
[f_SO4]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.0060359986852517
[]
[f_HCO3]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 5.0897287594019e-05
[]
[f_SiO2aq]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 3.0246609868421e-05
[]
[f_Al]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 3.268028901929e-08
[]
[f_Ca]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.00082159428184586
[]
[f_Mg]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.8546347062146e-05
[]
[f_Fe]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 4.3291908204093e-05
[]
[f_K]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 6.8434768308898e-05
[]
[f_Na]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.033298053919671
[]
[f_Sr]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.2771866652177e-05
[]
[f_F]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 5.5648860174073e-06
[]
[f_BOH]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.0003758574621917
[]
[f_Br]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 9.0315286107068e-05
[]
[f_Ba]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.5637460875161e-07
[]
[f_Li]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 8.3017067912701e-05
[]
[f_NO3]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.00010958455036169
[]
[f_O2aq]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = -7.0806852373351e-05
[]
[porepressure]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 30E6
[]
[temperature]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 92
scaling<<<{"description": "Specifies a scaling factor to apply to this variable"}>>> = 1E-6 # fluid enthalpy is roughly 1E6
[]
[]
[DiracKernels<<<{"href": "../../../syntax/DiracKernels/index.html"}>>>]
[inject_H]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_H
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_H
[]
[inject_Cl]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Cl
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Cl
[]
[inject_SO4]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_SO4
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_SO4
[]
[inject_HCO3]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_HCO3
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_HCO3
[]
[inject_SiO2aq]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_SiO2aq
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_SiO2aq
[]
[inject_Al]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Al
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Al
[]
[inject_Ca]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Ca
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Ca
[]
[inject_Mg]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Mg
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Mg
[]
[inject_Fe]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Fe
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Fe
[]
[inject_K]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_K
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_K
[]
[inject_Na]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Na
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Na
[]
[inject_Sr]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Sr
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Sr
[]
[inject_F]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_F
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_F
[]
[inject_BOH]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_BOH
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_BOH
[]
[inject_Br]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Br
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Br
[]
[inject_Ba]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Ba
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Ba
[]
[inject_Li]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_Li
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Li
[]
[inject_NO3]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_NO3
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_NO3
[]
[inject_O2aq]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_O2aq
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_O2aq
[]
[inject_H2O]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = injected_mass
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${injection_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
multiplying_var<<<{"description": "Fluxes will be moultiplied by this variable"}>>> = injection_rate_massfrac_H2O
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = injection.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = porepressure
[]
[produce_H]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_H
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 0
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_H
[]
[produce_Cl]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Cl
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 1
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Cl
[]
[produce_SO4]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_SO4
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 2
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_SO4
[]
[produce_HCO3]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_HCO3
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 3
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_HCO3
[]
[produce_SiO2aq]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_SiO2aq
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 4
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_SiO2aq
[]
[produce_Al]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Al
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 5
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Al
[]
[produce_Ca]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Ca
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 6
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Ca
[]
[produce_Mg]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Mg
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 7
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Mg
[]
[produce_Fe]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Fe
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 8
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Fe
[]
[produce_K]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_K
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 9
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_K
[]
[produce_Na]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Na
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 10
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Na
[]
[produce_Sr]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Sr
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 11
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Sr
[]
[produce_F]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_F
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 12
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_F
[]
[produce_BOH]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_BOH
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 13
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_BOH
[]
[produce_Br]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Br
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 14
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Br
[]
[produce_Ba]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Ba
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 15
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Ba
[]
[produce_Li]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_Li
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 16
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_Li
[]
[produce_NO3]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_NO3
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 17
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_NO3
[]
[produce_O2aq]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_O2aq
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 18
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = f_O2aq
[]
[produce_H2O]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_mass_H2O
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
mass_fraction_component<<<{"description": "The index corresponding to a fluid component. If supplied, the flux will be multiplied by the nodal mass fraction for the component"}>>> = 19
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = porepressure
[]
[produce_heat]
type = PorousFlowPolyLineSink<<<{"description": "Approximates a polyline sink by using a number of point sinks with given weighting whose positions are read from a file. NOTE: if you are using PorousFlowPorosity that depends on volumetric strain, you should set strain_at_nearest_qp=true in your GlobalParams, to ensure the nodal Porosity Material uses the volumetric strain at the Dirac quadpoints, and can therefore be computed", "href": "../../../source/dirackernels/PorousFlowPolyLineSink.html"}>>>
SumQuantityUO<<<{"description": "User Object of type=PorousFlowSumQuantity in which to place the total outflow from the line sink for each time step."}>>> = produced_heat
fluxes<<<{"description": "Tuple of flux values (measured in kg.m^-1.s^-1 if no 'use_*' are employed). These flux values are multiplied by the line-segment length to achieve a flux in kg.s^-1. A piecewise-linear fit is performed to the (p_or_t_vals,flux) pairs to obtain the flux at any arbitrary pressure (or temperature). If a quad-point pressure is less than the first pressure value, the first flux value is used. If quad-point pressure exceeds the final pressure value, the final flux value is used. This flux is OUT of the medium: hence positive values of flux means this will be a SINK, while negative values indicate this flux will be a SOURCE."}>>> = ${production_rate}
p_or_t_vals<<<{"description": "Tuple of pressure (or temperature) values. Must be monotonically increasing."}>>> = 0.0
use_enthalpy<<<{"description": "Multiply the flux by the fluid enthalpy"}>>> = true
point_file<<<{"description": "The file containing the coordinates of the points and their weightings that approximate the line sink. The physical meaning of the weightings depend on the scenario, eg, they may be borehole radii. Each line in the file must contain a space-separated weight and coordinate, viz r x y z. For boreholes, the last point in the file is defined as the borehole bottom, where the borehole pressure is bottom_pressure. If your file contains just one point, you must also specify the line_length and line_direction parameters. Note that you will get segementation faults if your points do not lie within your mesh!"}>>> = production.bh
variable<<<{"description": "The name of the variable that this residual object operates on"}>>> = temperature
[]
[]
[UserObjects<<<{"href": "../../../syntax/UserObjects/index.html"}>>>]
[injected_mass]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_H]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Cl]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_SO4]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_HCO3]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_SiO2aq]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Al]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Ca]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Mg]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Fe]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_K]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Na]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Sr]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_F]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_BOH]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Br]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Ba]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_Li]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_NO3]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_O2aq]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_mass_H2O]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[produced_heat]
type = PorousFlowSumQuantity<<<{"description": "Records total mass flowing into a borehole", "href": "../../../source/userobjects/PorousFlowSumQuantity.html"}>>>
[]
[]
[Postprocessors<<<{"href": "../../../syntax/Postprocessors/index.html"}>>>]
[dt]
type = TimestepSize<<<{"description": "Reports the timestep size", "href": "../../../source/postprocessors/TimestepSize.html"}>>>
execute_on<<<{"description": "The list of flag(s) indicating when this object should be executed. For a description of each flag, see https://mooseframework.inl.gov/source/interfaces/SetupInterface.html."}>>> = TIMESTEP_BEGIN
[]
[tot_kg_injected_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = injected_mass
[]
[kg_H_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_H
[]
[kg_Cl_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Cl
[]
[kg_SO4_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_SO4
[]
[kg_HCO3_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_HCO3
[]
[kg_SiO2aq_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_SiO2aq
[]
[kg_Al_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Al
[]
[kg_Ca_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Ca
[]
[kg_Mg_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Mg
[]
[kg_Fe_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Fe
[]
[kg_K_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_K
[]
[kg_Na_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Na
[]
[kg_Sr_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Sr
[]
[kg_F_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_F
[]
[kg_BOH_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_BOH
[]
[kg_Br_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Br
[]
[kg_Ba_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Ba
[]
[kg_Li_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_Li
[]
[kg_NO3_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_NO3
[]
[kg_O2aq_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_O2aq
[]
[kg_H2O_produced_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_mass_H2O
[]
[mole_rate_H_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_H
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_H_produced_this_timestep dt'
[]
[mole_rate_Cl_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Cl
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Cl_produced_this_timestep dt'
[]
[mole_rate_SO4_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_SO4
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_SO4_produced_this_timestep dt'
[]
[mole_rate_HCO3_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_HCO3
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_HCO3_produced_this_timestep dt'
[]
[mole_rate_SiO2aq_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_SiO2aq
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_SiO2aq_produced_this_timestep dt'
[]
[mole_rate_Al_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Al
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Al_produced_this_timestep dt'
[]
[mole_rate_Ca_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Ca
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Ca_produced_this_timestep dt'
[]
[mole_rate_Mg_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Mg
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Mg_produced_this_timestep dt'
[]
[mole_rate_Fe_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Fe
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Fe_produced_this_timestep dt'
[]
[mole_rate_K_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_K
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_K_produced_this_timestep dt'
[]
[mole_rate_Na_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Na
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Na_produced_this_timestep dt'
[]
[mole_rate_Sr_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Sr
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Sr_produced_this_timestep dt'
[]
[mole_rate_F_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_F
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_F_produced_this_timestep dt'
[]
[mole_rate_BOH_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_BOH
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_BOH_produced_this_timestep dt'
[]
[mole_rate_Br_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Br
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Br_produced_this_timestep dt'
[]
[mole_rate_Ba_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Ba
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Ba_produced_this_timestep dt'
[]
[mole_rate_Li_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_Li
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_Li_produced_this_timestep dt'
[]
[mole_rate_NO3_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_NO3
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_NO3_produced_this_timestep dt'
[]
[mole_rate_O2aq_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_O2aq
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_O2aq_produced_this_timestep dt'
[]
[mole_rate_H2O_produced]
type = FunctionValuePostprocessor<<<{"description": "Computes the value of a supplied function at a single point (scalable)", "href": "../../../source/postprocessors/FunctionValuePostprocessor.html"}>>>
function<<<{"description": "The function which supplies the postprocessor value."}>>> = moles_H2O
indirect_dependencies<<<{"description": "If the evaluated function depends on other postprocessors they must be listed here to ensure proper dependency resolution"}>>> = 'kg_H2O_produced_this_timestep dt'
[]
[heat_joules_extracted_this_timestep]
type = PorousFlowPlotQuantity<<<{"description": "Extracts the value from the PorousFlowSumQuantity UserObject", "href": "../../../source/postprocessors/PorousFlowPlotQuantity.html"}>>>
uo<<<{"description": "PorousFlowSumQuantity user object name that holds the required information"}>>> = produced_heat
[]
[production_temperature]
type = AverageNodalVariableValue<<<{"description": "Computes the average value of a field by sampling all nodal solutions on the domain or within a subdomain", "href": "../../../source/postprocessors/AverageNodalVariableValue.html"}>>>
boundary<<<{"description": "The list of boundaries (ids or names) from the mesh where this object applies"}>>> = production_nodes
variable<<<{"description": "The name of the variable that this postprocessor operates on"}>>> = temperature
[]
[]
[Functions<<<{"href": "../../../syntax/Functions/index.html"}>>>]
[moles_H]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_H dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_H_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_H * 1000 / 1.0079 / dt'
[]
[moles_Cl]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Cl dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Cl_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Cl * 1000 / 35.453 / dt'
[]
[moles_SO4]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_SO4 dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_SO4_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_SO4 * 1000 / 96.0576 / dt'
[]
[moles_HCO3]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_HCO3 dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_HCO3_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_HCO3 * 1000 / 61.0171 / dt'
[]
[moles_SiO2aq]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_SiO2aq dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_SiO2aq_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_SiO2aq * 1000 / 60.0843 / dt'
[]
[moles_Al]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Al dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Al_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Al * 1000 / 26.9815 / dt'
[]
[moles_Ca]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Ca dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Ca_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Ca * 1000 / 40.08 / dt'
[]
[moles_Mg]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Mg dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Mg_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Mg * 1000 / 24.305 / dt'
[]
[moles_Fe]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Fe dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Fe_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Fe * 1000 / 55.847 / dt'
[]
[moles_K]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_K dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_K_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_K * 1000 / 39.0983 / dt'
[]
[moles_Na]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Na dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Na_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Na * 1000 / 22.9898 / dt'
[]
[moles_Sr]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Sr dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Sr_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Sr * 1000 / 87.62 / dt'
[]
[moles_F]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_F dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_F_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_F * 1000 / 18.9984 / dt'
[]
[moles_BOH]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_BOH dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_BOH_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_BOH * 1000 / 61.8329 / dt'
[]
[moles_Br]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Br dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Br_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Br * 1000 / 79.904 / dt'
[]
[moles_Ba]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Ba dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Ba_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Ba * 1000 / 137.33 / dt'
[]
[moles_Li]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_Li dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_Li_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_Li * 1000 / 6.941 / dt'
[]
[moles_NO3]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_NO3 dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_NO3_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_NO3 * 1000 / 62.0049 / dt'
[]
[moles_O2aq]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_O2aq dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_O2aq_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_O2aq * 1000 / 31.9988 / dt'
[]
[moles_H2O]
type = ParsedFunction<<<{"description": "Function created by parsing a string", "href": "../../../source/functions/MooseParsedFunction.html"}>>>
symbol_names<<<{"description": "Symbols (excluding t,x,y,z) that are bound to the values provided by the corresponding items in the symbol_values vector."}>>> = 'kg_H2O dt'
symbol_values<<<{"description": "Constant numeric values, postprocessor names, function names, and scalar variables corresponding to the symbols in symbol_names."}>>> = 'kg_H2O_produced_this_timestep dt'
expression<<<{"description": "The user defined function."}>>> = 'kg_H2O * 1000 / 18.01801802 / dt'
[]
[]
[AuxVariables<<<{"href": "../../../syntax/AuxVariables/index.html"}>>>]
[injection_temperature]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 92
[]
[injection_rate_massfrac_H]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = -2.952985071156e-06
[]
[injection_rate_massfrac_Cl]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.04870664551708
[]
[injection_rate_massfrac_SO4]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.0060359986852517
[]
[injection_rate_massfrac_HCO3]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 5.0897287594019e-05
[]
[injection_rate_massfrac_SiO2aq]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 3.0246609868421e-05
[]
[injection_rate_massfrac_Al]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 3.268028901929e-08
[]
[injection_rate_massfrac_Ca]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.00082159428184586
[]
[injection_rate_massfrac_Mg]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.8546347062146e-05
[]
[injection_rate_massfrac_Fe]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 4.3291908204093e-05
[]
[injection_rate_massfrac_K]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 6.8434768308898e-05
[]
[injection_rate_massfrac_Na]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.033298053919671
[]
[injection_rate_massfrac_Sr]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.2771866652177e-05
[]
[injection_rate_massfrac_F]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 5.5648860174073e-06
[]
[injection_rate_massfrac_BOH]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.0003758574621917
[]
[injection_rate_massfrac_Br]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 9.0315286107068e-05
[]
[injection_rate_massfrac_Ba]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 1.5637460875161e-07
[]
[injection_rate_massfrac_Li]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 8.3017067912701e-05
[]
[injection_rate_massfrac_NO3]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.00010958455036169
[]
[injection_rate_massfrac_O2aq]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = -7.0806852373351e-05
[]
[injection_rate_massfrac_H2O]
initial_condition<<<{"description": "Specifies a constant initial condition for this variable"}>>> = 0.91032275033842
[]
[rate_H]
[]
[rate_Cl]
[]
[rate_SO4]
[]
[rate_HCO3]
[]
[rate_SiO2aq]
[]
[rate_Al]
[]
[rate_Ca]
[]
[rate_Mg]
[]
[rate_Fe]
[]
[rate_K]
[]
[rate_Na]
[]
[rate_Sr]
[]
[rate_F]
[]
[rate_BOH]
[]
[rate_Br]
[]
[rate_Ba]
[]
[rate_Li]
[]
[rate_NO3]
[]
[rate_O2aq]
[]
[rate_H2O]
[]
[]
[MultiApps<<<{"href": "../../../syntax/MultiApps/index.html"}>>>]
[react]
type = TransientMultiApp<<<{"description": "MultiApp for performing coupled simulations with the parent and sub-application both progressing in time.", "href": "../../../source/multiapps/TransientMultiApp.html"}>>>
input_files<<<{"description": "The input file for each App. If this parameter only contains one input file it will be used for all of the Apps. When using 'positions_from_file' it is also admissable to provide one input_file per file."}>>> = aquifer_geochemistry.i
clone_master_mesh<<<{"description": "True to clone parent app mesh and use it for this MultiApp."}>>> = true
execute_on<<<{"description": "The list of flag(s) indicating when this object should be executed. For a description of each flag, see https://mooseframework.inl.gov/source/interfaces/SetupInterface.html."}>>> = 'timestep_end'
[]
[]
[Transfers<<<{"href": "../../../syntax/Transfers/index.html"}>>>]
[changes_due_to_flow]
type = MultiAppCopyTransfer<<<{"description": "Copies variables (nonlinear and auxiliary) between multiapps that have identical meshes.", "href": "../../../source/transfers/MultiAppCopyTransfer.html"}>>>
source_variable<<<{"description": "The variable to transfer from."}>>> = 'rate_H rate_Cl rate_SO4 rate_HCO3 rate_SiO2aq rate_Al rate_Ca rate_Mg rate_Fe rate_K rate_Na rate_Sr rate_F rate_BOH rate_Br rate_Ba rate_Li rate_NO3 rate_O2aq rate_H2O temperature'
variable<<<{"description": "The auxiliary variable to store the transferred values in."}>>> = 'pf_rate_H pf_rate_Cl pf_rate_SO4 pf_rate_HCO3 pf_rate_SiO2aq pf_rate_Al pf_rate_Ca pf_rate_Mg pf_rate_Fe pf_rate_K pf_rate_Na pf_rate_Sr pf_rate_F pf_rate_BOH pf_rate_Br pf_rate_Ba pf_rate_Li pf_rate_NO3 pf_rate_O2aq pf_rate_H2O temperature'
to_multi_app<<<{"description": "The name of the MultiApp to transfer the data to"}>>> = react
[]
[massfrac_from_geochem]
type = MultiAppCopyTransfer<<<{"description": "Copies variables (nonlinear and auxiliary) between multiapps that have identical meshes.", "href": "../../../source/transfers/MultiAppCopyTransfer.html"}>>>
source_variable<<<{"description": "The variable to transfer from."}>>> = 'massfrac_H massfrac_Cl massfrac_SO4 massfrac_HCO3 massfrac_SiO2aq massfrac_Al massfrac_Ca massfrac_Mg massfrac_Fe massfrac_K massfrac_Na massfrac_Sr massfrac_F massfrac_BOH massfrac_Br massfrac_Ba massfrac_Li massfrac_NO3 massfrac_O2aq '
variable<<<{"description": "The auxiliary variable to store the transferred values in."}>>> = 'f_H f_Cl f_SO4 f_HCO3 f_SiO2aq f_Al f_Ca f_Mg f_Fe f_K f_Na f_Sr f_F f_BOH f_Br f_Ba f_Li f_NO3 f_O2aq '
from_multi_app<<<{"description": "The name of the MultiApp to receive data from"}>>> = react
[]
[]The interface with the child geochemistry simulation is created using a MultiApp and Transfers, which
provide the source-species rates to the
geochemistrysimulationupdate the mass fractions of each transported species using the results of the
geochemistrysimulation
[MultiApps<<<{"href": "../../../syntax/MultiApps/index.html"}>>>]
[react]
type = TransientMultiApp<<<{"description": "MultiApp for performing coupled simulations with the parent and sub-application both progressing in time.", "href": "../../../source/multiapps/TransientMultiApp.html"}>>>
input_files<<<{"description": "The input file for each App. If this parameter only contains one input file it will be used for all of the Apps. When using 'positions_from_file' it is also admissable to provide one input_file per file."}>>> = aquifer_geochemistry.i
clone_master_mesh<<<{"description": "True to clone parent app mesh and use it for this MultiApp."}>>> = true
execute_on<<<{"description": "The list of flag(s) indicating when this object should be executed. For a description of each flag, see https://mooseframework.inl.gov/source/interfaces/SetupInterface.html."}>>> = 'timestep_end'
[]
[][Transfers<<<{"href": "../../../syntax/Transfers/index.html"}>>>]
[changes_due_to_flow]
type = MultiAppCopyTransfer<<<{"description": "Copies variables (nonlinear and auxiliary) between multiapps that have identical meshes.", "href": "../../../source/transfers/MultiAppCopyTransfer.html"}>>>
source_variable<<<{"description": "The variable to transfer from."}>>> = 'rate_H rate_Cl rate_SO4 rate_HCO3 rate_SiO2aq rate_Al rate_Ca rate_Mg rate_Fe rate_K rate_Na rate_Sr rate_F rate_BOH rate_Br rate_Ba rate_Li rate_NO3 rate_O2aq rate_H2O temperature'
variable<<<{"description": "The auxiliary variable to store the transferred values in."}>>> = 'pf_rate_H pf_rate_Cl pf_rate_SO4 pf_rate_HCO3 pf_rate_SiO2aq pf_rate_Al pf_rate_Ca pf_rate_Mg pf_rate_Fe pf_rate_K pf_rate_Na pf_rate_Sr pf_rate_F pf_rate_BOH pf_rate_Br pf_rate_Ba pf_rate_Li pf_rate_NO3 pf_rate_O2aq pf_rate_H2O temperature'
to_multi_app<<<{"description": "The name of the MultiApp to transfer the data to"}>>> = react
[]
[massfrac_from_geochem]
type = MultiAppCopyTransfer<<<{"description": "Copies variables (nonlinear and auxiliary) between multiapps that have identical meshes.", "href": "../../../source/transfers/MultiAppCopyTransfer.html"}>>>
source_variable<<<{"description": "The variable to transfer from."}>>> = 'massfrac_H massfrac_Cl massfrac_SO4 massfrac_HCO3 massfrac_SiO2aq massfrac_Al massfrac_Ca massfrac_Mg massfrac_Fe massfrac_K massfrac_Na massfrac_Sr massfrac_F massfrac_BOH massfrac_Br massfrac_Ba massfrac_Li massfrac_NO3 massfrac_O2aq '
variable<<<{"description": "The auxiliary variable to store the transferred values in."}>>> = 'f_H f_Cl f_SO4 f_HCO3 f_SiO2aq f_Al f_Ca f_Mg f_Fe f_K f_Na f_Sr f_F f_BOH f_Br f_Ba f_Li f_NO3 f_O2aq '
from_multi_app<<<{"description": "The name of the MultiApp to receive data from"}>>> = react
[]
[]The injection and production is performed by PorousFlowPolyLinkSinks, for instance
[inject_H]
type = PorousFlowPolyLineSink
SumQuantityUO = injected_mass
fluxes = ${injection_rate}
p_or_t_vals = 0.0
multiplying_var = injection_rate_massfrac_H
point_file = injection.bh
variable = f_H
[]In this block, the injection_rate_massfrac_H is set to the initial mass fraction, representing pumping unadulterated reservoir water into the system
[injection_rate_massfrac_H]
initial_condition = -2.952985071156e-06
[]but will eventually be sourced from the parent simulation exchanger.i (see below). The production DiracKernels record the produced mass of each species into Postprocessors for interface with exchanger.i:
[produce_H]
type = PorousFlowPolyLineSink
SumQuantityUO = produced_mass_H
fluxes = ${production_rate}
p_or_t_vals = 0.0
mass_fraction_component = 0
point_file = production.bh
variable = f_H
[]and this is converted to a mole rate using Functions such as:
[moles_H]
type = ParsedFunction
symbol_names = 'kg_H dt'
symbol_values = 'kg_H_produced_this_timestep dt'
expression = 'kg_H * 1000 / 1.0079 / dt'
[]The heat exchanger
The heat exchanger simulation accepts produced water from the porous_flow simulation, heats it to 160C allowing precipitates to form and removing them, then injects the remaining fluid back to the porous_flow simulation. Its core is the TimeDependentReactionSolver:
the first lines are similar to previous input files, but notice that because
mode = 4("exchanger" mode) all the initial fluid is removed before the exchanger starts precipitating fluid from theporous_flowsimulationthe output temperature is controlled by the
ramp_temperatureAuxVariable, which ramps to 160C over approximately 1 day to allow theaquifer_geochemistrysimulation to easily convergethe
source_speciesrates are provided by theporous_flowsimulation
[TimeDependentReactionSolver<<<{"href": "../../../syntax/TimeDependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
include_moose_solve<<<{"description": "Include a usual MOOSE solve involving Variables and Kernels. In pure reaction systems (without transport) include_moose_solve = false is appropriate, but with transport 'true' must be used"}>>> = false
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = 'NO3- O2(aq)'
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = ' NH3 HS-'
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = 'Cl-'
# initial conditions are unimportant because in exchanger mode all existing fluid is flushed from the system before adding the produced water
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = 'H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NH3 HS-'
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = '1.0 1E-6 1E-6 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18'
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = 'kg_solvent_water bulk_composition bulk_composition free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration free_concentration'
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg moles moles molal molal molal molal molal molal molal molal molal molal molal molal molal molal molal molal molal"
prevent_precipitation<<<{"description": "Mineral species in this list will be prevented from precipitating, irrespective of their saturation index (unless they are initially in the basis)"}>>> = 'Fluorite Albite Goethite'
initial_temperature<<<{"description": "The initial aqueous solution is equilibrated at this system before adding reactants, changing temperature, etc."}>>> = 92
mode<<<{"description": "This may vary temporally. If mode=1 then 'dump' mode is used, which means all non-kinetic mineral masses are removed from the system before the equilibrium solution is sought (ie, removal occurs at the beginning of the time step). If mode=2 then 'flow-through' mode is used, which means all mineral masses are removed from the system after it the equilbrium solution has been found (ie, at the end of a time step). If mode=3 then 'flush' mode is used, then before the equilibrium solution is sought (ie, at the start of a time step) water+species is removed from the system at the same rate as pure water + non-mineral solutes are entering the system (specified in source_species_rates). If mode=4 then 'heat-exchanger' mode is used, which means the entire current aqueous solution is removed, then the source_species are added, then the temperature is set to 'cold_temperature', the system is solved and any precipitated minerals are removed, then the temperature is set to 'temperature', the system re-solved and any precipitated minerals are removed. If mode is any other number, no special mode is active (the system simply responds to the source_species_rates, controlled_activity_value, etc)."}>>> = 4
temperature<<<{"description": "Temperature. This has two different meanings if mode!=4. (1) If no species are being added to the solution (no source_species_rates are positive) then this is the temperature of the aqueous solution. (2) If species are being added, this is the temperature of the species being added. In case (2), the final aqueous-solution temperature is computed assuming the species are added, temperature is equilibrated and then, if species are also being removed, they are removed. If you wish to add species and simultaneously alter the temperature, you will have to use a sequence of heat-add-heat-add, etc steps. In the case that mode=4, temperature is the final temperature of the aqueous solution"}>>> = ramp_temperature # ramp up to 160degC over ~1 day so that aquifer geochemistry simulation can easily converge
cold_temperature<<<{"description": "This is only used if mode=4, where it is the cold temperature of the heat exchanger."}>>> = 92
heating_increments<<<{"description": "This is only used if mode=4. Internal to this object, the temperature is ramped from cold_temperature to temperature in heating_increments increments. This helps difficult problems converge"}>>> = 10
source_species_names<<<{"description": "The name of the species that are added at rates given in source_species_rates. There must be an equal number of these as source_species_rates."}>>> = ' H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq) H2O'
source_species_rates<<<{"description": "Rates, in mols/time_unit, of addition of the species with names given in source_species_names. A negative value corresponds to removing a species: be careful that you don't cause negative mass problems!"}>>> = ' production_rate_H production_rate_Cl production_rate_SO4 production_rate_HCO3 production_rate_SiO2aq production_rate_Al production_rate_Ca production_rate_Mg production_rate_Fe production_rate_K production_rate_Na production_rate_Sr production_rate_F production_rate_BOH production_rate_Br production_rate_Ba production_rate_Li production_rate_NO3 production_rate_O2aq production_rate_H2O'
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # max_ionic_strength in such a simple problem does not need ramping
[]The source_species_rates are provided by the porous_flow simulation using a Transfer:
[production_H]
type = MultiAppPostprocessorInterpolationTransfer
direction = FROM_MULTIAPP
multi_app = porous_flow_sim
postprocessor = mole_rate_H_produced
variable = production_rate_H
[]The composition of fluid injected from the exchanger to the porous_flow simulation depends on the transported composition:
[transported_H_auxk]
type = GeochemistryQuantityAux
quantity = transported_moles_in_original_basis
variable = transported_H
species = 'H+'
[]which is converted to a mass fraction:
[massfrac_H_auxk]
type = ParsedAux
coupled_variables = 'transported_mass transported_H'
variable = massfrac_H
expression = '1.0079 * transported_H / transported_mass'
[]before passing to the porous_flow simulation:
[injection_H]
type = MultiAppNearestNodeTransfer
direction = TO_MULTIAPP
multi_app = porous_flow_sim
fixed_meshes = true
source_variable = 'massfrac_H'
variable = 'injection_rate_massfrac_H'
[]A similar Transfer is used for the temperature of the injected fluid.
The amount of precipitate of each mineral is recorded using a GeochemistryQuantityAux:
[dumped_Siderite_auxk]
type = GeochemistryQuantityAux
variable = dumped_Siderite
species = Siderite
quantity = moles_dumped
[]Writing the input files
The input files for the aqueous geochemistry simulation, the porous flow simulation and the heat exchanger simulation are all quite lengthy and repetitive, simply because of the large number of species involved. To avoid typos, a python script is provided to write these input files:
# The purpose of this script is to create the input files named below
# The input files are conceptually fairly simple, but are quite long and repetitive because of the large number of species involved, so this python file helps prevent typos
porous_flow_filename = "porous_flow.i"
injection_bh_filename = "injection.bh"
production_bh_filename = "production.bh"
aquifer_geochem_filename = "aquifer_geochemistry.i"
exchanger_filename = "exchanger.i"
inject_rate = 0.02 # injection rate in kg/s/m
# names of the species in the porous-flow simulations (no brackets, minus signs, etc, so they can be used in Parsed quantities)
var_name = [
"H",
"Cl",
"SO4",
"HCO3",
"SiO2aq",
"Al",
"Ca",
"Mg",
"Fe",
"K",
"Na",
"Sr",
"F",
"BOH",
"Br",
"Ba",
"Li",
"NO3",
"O2aq",
"H2O",
]
# names of the geochemical species corresponding to var_name
geochem_vars = [
"H+",
"Cl-",
"SO4--",
"HCO3-",
"SiO2(aq)",
"Al+++",
"Ca++",
"Mg++",
"Fe++",
"K+",
"Na+",
"Sr++",
"F-",
"B(OH)3",
"Br-",
"Ba++",
"Li+",
"NO3-",
"O2(aq)",
"H2O",
]
# these are the initial mass fractions of the var_name species. The numbers are obtained by recording the transported mass fractions of each species as postprocessors: run one time-step of aquifer_geochemistry.i to find the numbers
ic = [
-2.952985071156e-06,
0.04870664551708,
0.0060359986852517,
5.0897287594019e-05,
3.0246609868421e-05,
3.268028901929e-08,
0.00082159428184586,
1.8546347062146e-05,
4.3291908204093e-05,
6.8434768308898e-05,
0.033298053919671,
1.2771866652177e-05,
5.5648860174073e-06,
0.0003758574621917,
9.0315286107068e-05,
1.5637460875161e-07,
8.3017067912701e-05,
0.00010958455036169,
-7.0806852373351e-05,
0.91032275033842,
]
# mol weight of each species
mol_weight = [
1.0079,
35.453,
96.0576,
61.0171,
60.0843,
26.9815,
40.08,
24.305,
55.847,
39.0983,
22.9898,
87.62,
18.9984,
61.8329,
79.904,
137.33,
6.941,
62.0049,
31.9988,
18.01801802,
]
# all the minerals in the system
all_minerals = [
"Siderite",
"Pyrrhotite",
"Dolomite",
"Illite",
"Anhydrite",
"Calcite",
"Quartz",
"K-feldspar",
"Kaolinite",
"Barite",
"Celestite",
"Fluorite",
"Albite",
"Chalcedony",
"Goethite",
]
# this impacts the Mesh created and the borehole Dirac points
# resolution = 1 means lowest resolution
# resolution = 2 is higher
resolution = 1
def write_header(f):
# all file written have this preprended to them to alert a reader that it wasn't written by hand
f.write("#########################################\n")
f.write("# #\n")
f.write("# File written by create_input_files.py #\n")
f.write("# #\n")
f.write("#########################################\n")
def write_executioner(f):
# executioner used
f.write(
"[Executioner]\n type = Transient\n solve_type = Newton\n end_time = 7.76E6 # 90 days\n"
)
f.write(
" [./TimeStepper]\n type = FunctionDT\n function = 'min(3E4, max(1E4, 0.2 * t))'\n [../]\n[]\n"
)
def nxnynz(res):
# return (nx, ny, nz) for the specified resolution. This is used in creating the mesh, and in the nodal_volume AuxKernel (until PR #15691 is merged)
# Note that if you make another "res" option that has a high nz, you'll also have to increase the number of z coords in injection_by_filename and production_bh_filename
nx = 15
ny = 4
nz = 5
if res == 2:
nx = 30
ny = 8
nz = 10
return (nx, ny, nz)
def write_mesh(f, res):
# 3D mesh used
f.write("[Mesh]\n [gen]\n type = GeneratedMeshGenerator\n dim = 3\n")
f.write(
" xmin = -75\n xmax = 75\n ymin = 0\n ymax = 40\n zmin = -25\n zmax = 25\n"
)
nx, ny, nz = nxnynz(res)
f.write(
" nx = " + str(nx) + "\n ny = " + str(ny) + "\n nz = " + str(nz) + "\n"
)
f.write(" []\n")
f.write(
" [./aquifer]\n type = ParsedSubdomainMeshGenerator\n input = gen\n block_id = 1\n block_name = aquifer\n combinatorial_geometry = 'z >= -5 & z <= 5'\n [../]\n"
)
# res = 1 means low-resolution
injection_nodesets = "'-25 0 -5; -25 0 5'"
production_nodesets = "'25 0 -5; 25 0 5'"
if res == 2:
injection_nodesets = "'-25 0 -5; -25 0 0; -25 0 5'"
production_nodesets = "'25 0 -5; 25 0 0; 25 0 5'"
f.write(
" [./injection_nodes]\n input = aquifer\n type = ExtraNodesetGenerator\n new_boundary = injection_nodes\n coord = "
+ injection_nodesets
+ "\n [../]\n"
)
f.write(
" [./production_nodes]\n input = injection_nodes\n type = ExtraNodesetGenerator\n new_boundary = production_nodes\n coord = "
+ production_nodesets
+ "\n [../]\n"
)
f.write("[]\n")
import os
import sys
sys.stdout.write(
"Outputting injection borehole specification to " + injection_bh_filename + "\n"
)
f = open(injection_bh_filename, "w")
write_header(f)
for z in [-5, -3, -1, 1, 3, 5]:
f.write("1.0 -25.0 0.0 " + str(z) + "\n")
f.close()
sys.stdout.write(
"Outputting production borehole specification to " + production_bh_filename + "\n"
)
f = open(production_bh_filename, "w")
write_header(f)
for z in [-5, -3, -1, 1, 3, 5]:
f.write("1.0 25.0 0.0 " + str(z) + "\n")
f.close()
sys.stdout.write("Outputting porous-flow input file to " + porous_flow_filename + "\n")
f = open(porous_flow_filename, "w")
write_header(f)
f.write(
"# PorousFlow simulation of injection and production in a simplified GeoTES aquifer\n# Much of this file is standard porous-flow stuff. The unusual aspects are:\n# - transfer of the rates of changes of each species (kg.s) to the aquifer_geochemistry.i simulation. This is achieved by saving these changes from the PorousFlowMassTimeDerivative residuals\n# - transfer of the temperature field to the aquifer_geochemistry.i simulation\n# Interesting behaviour can be simulated by this file without its 'parent' simulation, exchanger.i. exchanger.i provides mass-fractions injected via the injection_rate_massfrac_* variables, but since these are more-or-less constant throughout the duration of the exchanger.i simulation, the initial_conditions specified below may be used. Similar, exchanger.i provides injection_temperature, but that is also constant.\n"
)
f.write(
"injection_rate = "
+ str(-inject_rate)
+ " # kg/s/m, negative because injection as a source\n"
)
f.write(
"production_rate = "
+ str(inject_rate)
+ " # kg/s/m, this is about the maximum that can be sustained by the aquifer, with its fairly low permeability, without porepressure becoming negative\n"
)
write_mesh(f, resolution)
f.write(
"\n[GlobalParams]\n PorousFlowDictator = dictator\n gravity = '0 0 -10'\n[]\n"
)
f.write(
"[BCs]\n [./injection_temperature]\n type = MatchedValueBC\n variable = temperature\n v = injection_temperature\n boundary = injection_nodes\n [../]\n[]\n"
)
f.write(
"[Modules]\n [./FluidProperties]\n [./the_simple_fluid]\n type = SimpleFluidProperties\n thermal_expansion = 0\n bulk_modulus = 2E9\n viscosity = 1E-3\n density0 = 1000\n cv = 4000.0\n cp = 4000.0\n [../]\n [../]\n[]\n\n"
)
f.write(
"[Materials]\n [./temperature]\n type = PorousFlowTemperature\n temperature = temperature\n [../]\n [./fluid_props]\n type = PorousFlowSingleComponentFluid\n fp = the_simple_fluid\n temperature_unit = Celsius\n phase = 0\n [../]\n [./saturation]\n type = PorousFlow1PhaseP\n porepressure = porepressure\n capillary_pressure = capillary_pressure\n [../]\n [./relperm]\n type = PorousFlowRelativePermeabilityConst\n phase = 0\n [../]\n [./porosity_caps]\n type = PorousFlowPorosityConst # this simulation has no porosity changes from dissolution\n block = 0\n porosity = 0.01\n [../]\n [./porosity_aquifer]\n type = PorousFlowPorosityConst # this simulation has no porosity changes from dissolution\n block = aquifer\n porosity = 0.063\n [../]\n [./permeability_caps]\n type = PorousFlowPermeabilityConst\n block = 0\n permeability = '1E-18 0 0 0 1E-18 0 0 0 1E-18'\n [../]\n [./permeability_aquifer]\n type = PorousFlowPermeabilityConst\n block = aquifer\n permeability = '1.7E-15 0 0 0 1.7E-15 0 0 0 4.1E-16'\n [../]\n [./rock_heat]\n type = PorousFlowMatrixInternalEnergy\n density = 2500.0\n specific_heat_capacity = 1200.0\n [../]\n"
)
f.write(
" [./mass_frac]\n type = PorousFlowMassFraction\n mass_fraction_vars = '"
)
for i in range(19):
f.write("f_" + var_name[i] + " ")
f.write("'\n [../]\n")
f.write("[]\n")
f.write(
"[Preconditioning]\n active = typically_efficient\n [./typically_efficient]\n type = SMP\n full = true\n petsc_options_iname = '-pc_type -pc_hypre_type'\n petsc_options_value = ' hypre boomeramg'\n [../]\n [./strong]\n type = SMP\n full = true\n petsc_options = '-ksp_diagonal_scale -ksp_diagonal_scale_fix'\n petsc_options_iname = '-pc_type -sub_pc_type -sub_pc_factor_shift_type -pc_asm_overlap'\n petsc_options_value = ' asm ilu NONZERO 2'\n [../]\n [./probably_too_strong]\n type = SMP\n full = true\n petsc_options_iname = '-pc_type -pc_factor_mat_solver_package'\n petsc_options_value = ' lu mumps'\n [../]\n[]\n"
)
write_executioner(f)
f.write("[Outputs]\n exodus = true\n[]\n")
f.write("[Variables]\n")
for i in range(19):
f.write(
" [./f_"
+ var_name[i]
+ "]\n initial_condition = "
+ str(ic[i])
+ "\n [../]\n"
)
f.write(
" [./porepressure]\n initial_condition = 30E6\n [../]\n [./temperature]\n initial_condition = 92\n scaling = 1E-6 # fluid enthalpy is roughly 1E6\n [../]\n[]\n"
)
f.write("\n")
f.write("[DiracKernels]\n")
for i in range(19):
f.write(
" [./inject_"
+ var_name[i]
+ "]\n type = PorousFlowPolyLineSink\n SumQuantityUO = injected_mass\n fluxes = ${injection_rate}\n p_or_t_vals = 0.0\n multiplying_var = injection_rate_massfrac_"
+ var_name[i]
+ "\n point_file = "
+ injection_bh_filename
+ "\n variable = f_"
+ var_name[i]
+ "\n [../]\n"
)
i = 19
f.write(
" [./inject_"
+ var_name[i]
+ "]\n type = PorousFlowPolyLineSink\n SumQuantityUO = injected_mass\n fluxes = ${injection_rate}\n p_or_t_vals = 0.0\n multiplying_var = injection_rate_massfrac_"
+ var_name[i]
+ "\n point_file = "
+ injection_bh_filename
+ "\n variable = porepressure\n [../]\n"
)
f.write("\n")
for i in range(19):
f.write(
" [./produce_"
+ var_name[i]
+ "]\n type = PorousFlowPolyLineSink\n SumQuantityUO = produced_mass_"
+ var_name[i]
+ "\n fluxes = ${production_rate}\n p_or_t_vals = 0.0\n mass_fraction_component = "
+ str(i)
+ "\n point_file = "
+ production_bh_filename
+ "\n variable = f_"
+ var_name[i]
+ "\n [../]\n"
)
i = 19
f.write(
" [./produce_"
+ var_name[i]
+ "]\n type = PorousFlowPolyLineSink\n SumQuantityUO = produced_mass_"
+ var_name[i]
+ "\n fluxes = ${production_rate}\n p_or_t_vals = 0.0\n mass_fraction_component = "
+ str(i)
+ "\n point_file = "
+ production_bh_filename
+ "\n variable = porepressure\n [../]\n"
)
f.write(
" [./produce_heat]\n type = PorousFlowPolyLineSink\n SumQuantityUO = produced_heat\n fluxes = ${production_rate}\n p_or_t_vals = 0.0\n use_enthalpy = true\n point_file = "
+ production_bh_filename
+ "\n variable = temperature\n [../]\n"
)
f.write("[]\n")
f.write("\n")
f.write("[UserObjects]\n")
f.write(" [./injected_mass]\n type = PorousFlowSumQuantity\n [../]\n")
for i in range(20):
f.write(
" [./produced_mass_"
+ var_name[i]
+ "]\n type = PorousFlowSumQuantity\n [../]\n"
)
f.write(" [./produced_heat]\n type = PorousFlowSumQuantity\n [../]\n")
f.write("\n")
f.write(
" [./capillary_pressure]\n type = PorousFlowCapillaryPressureConst\n [../]\n"
)
f.write("\n")
f.write(
" [./dictator]\n type = PorousFlowDictator\n porous_flow_vars = 'porepressure temperature"
)
for i in range(19):
f.write(" f_" + var_name[i])
f.write(
"'\n number_fluid_phases = 1\n number_fluid_components = "
+ str(len(var_name))
+ "\n [../]\n"
)
f.write("[]")
f.write("\n")
f.write("[Postprocessors]\n")
f.write(
" [./dt]\n type = TimestepSize\n execute_on = TIMESTEP_BEGIN\n [../]\n [./tot_kg_injected_this_timestep]\n type = PorousFlowPlotQuantity\n uo = injected_mass\n [../]\n"
)
for i in range(20):
f.write(
" [./kg_"
+ var_name[i]
+ "_produced_this_timestep]\n type = PorousFlowPlotQuantity\n uo = produced_mass_"
+ var_name[i]
+ "\n [../]\n"
)
for i in range(20):
f.write(
" [./mole_rate_"
+ var_name[i]
+ "_produced]\n type = FunctionValuePostprocessor\n function = moles_"
+ var_name[i]
+ "\n [../]\n"
)
f.write(
" [./heat_joules_extracted_this_timestep]\n type = PorousFlowPlotQuantity\n uo = produced_heat\n [../]\n [./production_temperature]\n type = AverageNodalVariableValue\n boundary = production_nodes\n variable = temperature\n [../]\n"
)
f.write("[]\n")
f.write("\n")
f.write("[Functions]\n")
for i in range(20):
f.write(
" [./moles_"
+ var_name[i]
+ "]\n type = ParsedFunction\n vars = 'kg_"
+ var_name[i]
+ " dt'\n vals = 'kg_"
+ var_name[i]
+ "_produced_this_timestep dt'\n value = 'kg_"
+ var_name[i]
+ " * 1000 / "
+ str(mol_weight[i])
+ " / dt'\n [../]\n"
)
f.write("[]\n")
f.write("\n")
f.write("[Kernels]\n")
for i in range(19):
f.write(
" [./advective_flux_"
+ var_name[i]
+ "]\n type = PorousFlowAdvectiveFlux\n fluid_component = "
+ str(i)
+ "\n variable = f_"
+ var_name[i]
+ "\n [../]\n"
)
i = 19
f.write(
" [./advective_flux_"
+ var_name[i]
+ "]\n type = PorousFlowAdvectiveFlux\n fluid_component = "
+ str(i)
+ "\n variable = porepressure\n [../]\n"
)
for i in range(19):
f.write(
" [./time_deriv_"
+ var_name[i]
+ "]\n type = PorousFlowMassTimeDerivative\n fluid_component = "
+ str(i)
+ "\n save_in = rate_"
+ str(var_name[i])
+ " # change in kg at every node / dt\n variable = f_"
+ var_name[i]
+ "\n [../]\n"
)
i = 19
f.write(
" [./time_deriv_"
+ var_name[i]
+ "]\n type = PorousFlowMassTimeDerivative\n fluid_component = "
+ str(i)
+ "\n save_in = rate_"
+ str(var_name[i])
+ " # change in kg at every node / dt\n variable = porepressure\n [../]\n"
)
f.write(
" [./temperature_advection]\n type = PorousFlowHeatAdvection\n variable = temperature\n [../]\n [./temperature_time_deriv]\n type = PorousFlowEnergyTimeDerivative\n variable = temperature\n [../]\n"
)
f.write("[]\n")
f.write("\n")
f.write(
"[AuxVariables]\n [./injection_temperature]\n initial_condition = 92\n [../]\n"
)
for i in range(20):
f.write(
" [./injection_rate_massfrac_"
+ var_name[i]
+ "]\n initial_condition = "
+ str(ic[i])
+ "\n [../]\n"
)
for i in range(20):
f.write(" [./rate_" + var_name[i] + "]\n [../]\n")
f.write("[]\n")
f.write("\n")
f.write(
"[MultiApps]\n [./react]\n type = TransientMultiApp\n input_files = aquifer_geochemistry.i\n clone_master_mesh = true\n execute_on = 'timestep_end'\n [../]\n[]\n"
)
f.write("[Transfers]\n")
f.write(
" [./changes_due_to_flow]\n type = MultiAppCopyTransfer\n direction = to_multiapp\n source_variable = '"
)
for i in range(20):
f.write("rate_" + var_name[i] + " ")
f.write("temperature'\n")
f.write(" variable = '")
for i in range(20):
f.write("pf_rate_" + var_name[i] + " ")
f.write("temperature'\n")
f.write(" multi_app = react\n [../]\n")
f.write(
" [./massfrac_from_geochem]\n type = MultiAppCopyTransfer\n direction = from_multiapp\n source_variable = '"
)
for i in range(19):
f.write("massfrac_" + var_name[i] + " ")
f.write("'\n")
f.write(" variable = '")
for i in range(19):
f.write("f_" + var_name[i] + " ")
f.write("'\n multi_app = react\n [../]\n[]\n")
f.close()
sys.stdout.write(
"Outputting porous-flow input file to " + aquifer_geochem_filename + "\n"
)
f = open(aquifer_geochem_filename, "w")
write_header(f)
f.write(
"# Simulates geochemistry in the aquifer. This input file may be run in standalone fashion but it does not do anything of interest. To simulate something interesting, run the "
+ porous_flow_filename
+ " simulation which couples to this input file using MultiApps.\n"
)
f.write("# This file receives")
for i in range(20):
f.write(" pf_rate_" + var_name[i])
f.write(" and temperature as AuxVariables from " + porous_flow_filename + "\n")
f.write(
"# The pf_rate quantities are kg/s changes of fluid-component mass at each node, but the geochemistry module expects rates-of-changes of moles at every node. Secondly, since this input file considers just 1 litre of aqueous solution at every node, the nodal_void_volume is used to convert pf_rate_* into rate_*_per_1l, which is measured in mol/s/1_litre_of_aqueous_solution.\n"
)
f.write("# This file sends")
for i in range(19):
f.write(" massfrac_" + var_name[i])
f.write(
" to "
+ porous_flow_filename
+ ". These are computed from the corresponding transported_* quantities.\n"
)
f.write("\n")
f.write(
"[UserObjects]\n [./definition]\n type = GeochemicalModelDefinition\n database_file = '../../../../geochemistry/database/moose_geochemdb.json'\n basis_species = 'H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq)'\n equilibrium_minerals = '"
+ " ".join(all_minerals)
+ "'\n [../]\n [./nodal_void_volume_uo]\n type = NodalVoidVolume\n porosity = porosity\n execute_on = 'initial timestep_end' # initial means this is evaluated properly for the first timestep\n [../]\n[]\n"
)
f.write("\n")
f.write(
"[SpatialReactionSolver]\n model_definition = definition\n geochemistry_reactor_name = reactor\n"
)
f.write(" charge_balance_species = 'Cl-'\n")
f.write(
" swap_out_of_basis = 'NO3- H+ Fe++ Ba++ SiO2(aq) Mg++ O2(aq) Al+++ K+ Ca++ HCO3-'\n swap_into_basis = ' NH3 Pyrrhotite K-feldspar Barite Quartz Dolomite Siderite Calcite Illite Anhydrite Kaolinite'\n"
)
f.write("# ASSUME that 1 litre of solution contains:\n")
f.write(
" constraint_species = 'H2O Quartz Calcite K-feldspar Siderite Dolomite Anhydrite Pyrrhotite Illite Kaolinite Barite Na+ Cl- SO4-- Li+ B(OH)3 Br- F- Sr++ NH3'\n"
)
f.write(
" constraint_value = ' 0.99778351 322.177447 12.111108 6.8269499 6.2844304 2.8670301 1.1912027 0.51474767 0.3732507 0.20903322 0.0001865889 1.5876606 1.5059455 0.046792579 0.013110503 0.006663119 0.001238987 0.00032108 0.000159781 0.001937302'\n"
)
f.write(
" constraint_meaning = 'kg_solvent_water moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species moles_bulk_species'\n"
)
f.write(" prevent_precipitation = 'Fluorite Albite Goethite'\n")
f.write(" initial_temperature = 92\n")
f.write(" temperature = temperature\n")
f.write(
" source_species_names = 'H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq) H2O'\n"
)
f.write(" source_species_rates = '")
for i in range(20):
f.write(" rate_" + var_name[i] + "_per_1l")
f.write("'\n")
f.write(
" ramp_max_ionic_strength_initial = 0 # max_ionic_strength in such a simple problem does not need ramping\n"
)
f.write(
" execute_console_output_on = '' # only CSV and exodus output for this simulation\n"
)
f.write(
" add_aux_molal = false # save some memory and reduce variables in output exodus\n"
)
f.write(
" add_aux_mg_per_kg = false # save some memory and reduce variables in output exodus\n"
)
f.write(
" add_aux_free_mg = false # save some memory and reduce variables in output exodus\n"
)
f.write(
" add_aux_activity = false # save some memory and reduce variables in output exodus\n"
)
f.write(
" add_aux_bulk_moles = false # save some memory and reduce variables in output exodus\n"
)
f.write(" adaptive_timestepping = true\n")
f.write("[]\n")
f.write("\n")
write_mesh(f, resolution)
f.write("[GlobalParams]\n point = '-25 0 0'\n reactor = reactor\n[]\n")
write_executioner(f)
f.write(
"[AuxVariables]\n [./temperature]\n initial_condition = 92.0\n [../]\n [./porosity]\n initial_condition = 0.1\n [../]\n [./nodal_void_volume]\n [../]\n [./free_cm3_Kfeldspar] # necessary because of the minus sign in K-feldspar which does not parse correctly in the porosity AuxKernel\n [../]\n"
)
for i in range(20):
f.write(
" [./pf_rate_"
+ var_name[i]
+ "] # change in "
+ var_name[i]
+ " mass (kg/s) at each node provided by the porous-flow simulation\n [../]\n"
)
for i in range(20):
f.write(" [./rate_" + var_name[i] + "_per_1l]\n [../]\n")
for i in range(20):
f.write(" [./transported_" + var_name[i] + "]\n [../]\n")
f.write(" [./transported_mass]\n [../]\n")
for i in range(20):
f.write(" [./massfrac_" + var_name[i] + "]\n [../]\n")
f.write("[]\n")
f.write("\n")
f.write("[AuxKernels]\n")
f.write(
" [./free_cm3_Kfeldspar]\n type = GeochemistryQuantityAux\n variable = free_cm3_Kfeldspar\n species = 'K-feldspar'\n quantity = free_cm3\n execute_on = 'timestep_end'\n [../]\n"
)
f.write(
" [./porosity_auxk]\n type = ParsedAux\n args = 'free_cm3_Siderite free_cm3_Pyrrhotite free_cm3_Dolomite free_cm3_Illite free_cm3_Anhydrite free_cm3_Calcite free_cm3_Quartz free_cm3_Kfeldspar free_cm3_Kaolinite free_cm3_Barite free_cm3_Celestite free_cm3_Fluorite free_cm3_Albite free_cm3_Chalcedony free_cm3_Goethite'\n function = '1000.0 / (1000.0 + free_cm3_Siderite + free_cm3_Pyrrhotite + free_cm3_Dolomite + free_cm3_Illite + free_cm3_Anhydrite + free_cm3_Calcite + free_cm3_Quartz + free_cm3_Kfeldspar + free_cm3_Kaolinite + free_cm3_Barite + free_cm3_Celestite + free_cm3_Fluorite + free_cm3_Albite + free_cm3_Chalcedony + free_cm3_Goethite)'\n variable = porosity\n execute_on = 'timestep_end'\n [../]\n"
)
f.write(
" [./nodal_void_volume_auxk]\n type = NodalVoidVolumeAux\n variable = nodal_void_volume\n nodal_void_volume_uo = nodal_void_volume_uo\n execute_on = 'initial timestep_end' # initial to ensure it is properly evaluated for the first timestep\n [../]\n"
)
for i in range(20):
f.write(" [./rate_" + var_name[i] + "_per_1l_auxk]\n")
f.write(" type = ParsedAux\n")
f.write(" args = 'pf_rate_" + var_name[i] + " nodal_void_volume'\n")
f.write(" variable = rate_" + var_name[i] + "_per_1l\n")
f.write(
" function = 'pf_rate_"
+ var_name[i]
+ " / "
+ str(mol_weight[i])
+ " / nodal_void_volume'\n"
)
f.write(" execute_on = 'timestep_end'\n [../]\n")
for i in range(20):
f.write(" [./transported_" + var_name[i] + "_auxk]\n")
f.write(" type = GeochemistryQuantityAux\n")
f.write(" variable = transported_" + var_name[i] + "\n")
f.write(" species = '" + geochem_vars[i] + "'\n")
f.write(
" quantity = transported_moles_in_original_basis\n execute_on = 'timestep_end'\n [../]\n"
)
f.write(" [./transported_mass_auxk]\n type = ParsedAux\n args = '")
for i in range(20):
f.write(" transported_" + var_name[i])
f.write("'\n")
f.write(" variable = transported_mass\n")
f.write(" function = 'transported_" + var_name[0] + " * " + str(mol_weight[0]))
for i in range(1, 20):
f.write(" + transported_" + var_name[i] + " * " + str(mol_weight[i]))
f.write("'\n")
f.write(" execute_on = 'timestep_end'\n [../]\n")
for i in range(20):
f.write(" [./massfrac_" + var_name[i] + "_auxk]\n type = ParsedAux\n")
f.write(" args = 'transported_" + var_name[i] + " transported_mass'\n")
f.write(" variable = massfrac_" + var_name[i] + "\n")
f.write(
" function = 'transported_"
+ var_name[i]
+ " * "
+ str(mol_weight[i])
+ " / transported_mass'\n"
)
f.write(" execute_on = 'timestep_end'\n")
f.write(" [../]\n")
f.write("[]\n")
f.write("\n")
f.write("[Postprocessors]\n")
f.write(" [./memory]\n type = MemoryUsage\n outputs = 'console'\n [../]\n")
f.write(
" [./porosity]\n type = PointValue\n variable = porosity\n [../]\n [./solution_temperature]\n type = PointValue\n variable = solution_temperature\n [../]\n"
)
for i in range(20):
f.write(
" [./massfrac_"
+ var_name[i]
+ "]\n type = PointValue\n variable = massfrac_"
+ var_name[i]
+ "\n [../]\n"
)
for mineral in all_minerals:
f.write(
" [./free_cm3_"
+ mineral
+ "]\n type = PointValue\n variable = free_cm3_"
+ mineral
+ "\n [../]\n"
)
f.write("[]\n")
f.write("[Outputs]\n exodus = true\n csv = true\n[]\n")
f.close()
sys.stdout.write("Outputting heat-exchanger input file to " + exchanger_filename + "\n")
f = open(exchanger_filename, "w")
write_header(f)
f.write("# Model of the heat-exchanger\n")
f.write(
"# The input fluid to the heat exchanger is determined by AuxVariables called production_temperature"
)
for i in range(20):
f.write(", production_rate_" + var_name[i])
f.write(
". These come from Postprocessors in the "
+ porous_flow_filename
+ " simulation that measure the fluid composition at the production well.\n"
)
f.write(
"# Given the input fluid, the exchanger cools/heats the fluid, removing any precipitates, and injects fluid back to "
+ porous_flow_filename
+ " at temperature output_temperature and composition given by massfrac_H, etc.\n"
)
f.write("\n")
f.write(
"[UserObjects]\n [./definition]\n type = GeochemicalModelDefinition\n database_file = '../../../../geochemistry/database/moose_geochemdb.json'\n basis_species = 'H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NO3- O2(aq)'\n equilibrium_minerals = '"
+ " ".join(all_minerals)
+ "'\n [../]\n[]\n"
)
f.write("\n")
f.write(
"[TimeDependentReactionSolver]\n model_definition = definition\n include_moose_solve = false\n geochemistry_reactor_name = reactor\n"
)
f.write(" swap_out_of_basis = 'NO3- O2(aq)'\n swap_into_basis = ' NH3 HS-'\n")
f.write(" charge_balance_species = 'Cl-'\n")
f.write(
"# initial conditions are unimportant because in exchanger mode all existing fluid is flushed from the system before adding the produced water\n"
)
f.write(
" constraint_species = 'H2O H+ Cl- SO4-- HCO3- SiO2(aq) Al+++ Ca++ Mg++ Fe++ K+ Na+ Sr++ F- B(OH)3 Br- Ba++ Li+ NH3 HS-'\n"
)
f.write(
" constraint_value = '1.0 1E-6 1E-6 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18 1E-18'\n"
)
f.write(
" constraint_meaning = 'kg_solvent_water moles_bulk_species moles_bulk_species free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality free_molality'\n"
)
f.write(" prevent_precipitation = 'Fluorite Albite Goethite'\n")
f.write(" initial_temperature = 92\n")
f.write(" mode = 4\n")
f.write(
" temperature = ramp_temperature # ramp up to 160degC over ~1 day so that aquifer geochemistry simulation can easily converge\n"
)
f.write(" cold_temperature = 92\n")
f.write(" heating_increments = 10\n")
f.write(" source_species_names = '")
for i in range(20):
f.write(" " + geochem_vars[i])
f.write("'\n")
f.write(" source_species_rates = '")
for i in range(20):
f.write(" production_rate_" + var_name[i])
f.write("'\n")
f.write(
" ramp_max_ionic_strength_initial = 0 # max_ionic_strength in such a simple problem does not need ramping\n"
)
f.write("[]\n")
f.write("\n")
f.write("[GlobalParams]\n point = '0 0 0'\n reactor = reactor\n[]\n")
f.write("\n")
f.write("[AuxVariables]\n")
f.write(" [./ramp_temperature]\n initial_condition = 92\n [../]\n")
f.write(
" [./production_temperature]\n initial_condition = 92 # the production_T Transfer lags one timestep behind for some reason, so give this a reasonable initial condition\n [../]\n"
)
for i in range(20):
f.write(" [./transported_" + var_name[i] + "]\n [../]\n")
f.write(" [./transported_mass]\n [../]\n")
for i in range(20):
f.write(" [./massfrac_" + var_name[i] + "]\n [../]\n")
for mineral in all_minerals:
f.write(" [./dumped_" + mineral + "]\n [../]\n")
f.write(
"# The production_* Transfers lag one timestep behind for some reason (when the porous_flow simulation has finished, it correctly computes mole_rate_*_produced, but the Transfer gets the mole_rate_*_produced from the previous timestep), so give the production_rate_* reasonable initial conditions, otherwise they will be zero at the start of the simulation.\n"
)
for i in range(20):
f.write(" [./production_rate_" + var_name[i] + "]\n")
# mols/second = mass_fraction * inject_rate * aquifer_height * 1000 / mol_weight
f.write(
" initial_condition = "
+ str(ic[i] * inject_rate * 10 * 1000 / mol_weight[i])
+ "\n"
)
f.write(" [../]\n")
f.write("[]\n")
f.write("\n")
f.write("[AuxKernels]\n")
f.write(
" [./ramp_temperature]\n type = FunctionAux\n variable = ramp_temperature\n function = 'min(160, max(92, 92 + (160 - 92) * t / 1E5))'\n [../]\n"
)
for i in range(20):
f.write(" [./transported_" + var_name[i] + "_auxk]\n")
f.write(
" type = GeochemistryQuantityAux\n quantity = transported_moles_in_original_basis\n"
)
f.write(" variable = transported_" + var_name[i] + "\n")
f.write(" species = '" + geochem_vars[i] + "'\n")
f.write(" [../]\n")
f.write(" [./transported_mass_auxk]\n")
f.write(" type = ParsedAux\n")
f.write(" args = '")
for i in range(20):
f.write(" transported_" + var_name[i])
f.write("'\n")
f.write(" variable = transported_mass\n")
f.write(" function = '")
for i in range(19):
f.write(" transported_" + var_name[i] + " * " + str(mol_weight[i]) + " +")
i = 19
f.write(" transported_" + var_name[i] + " * " + str(mol_weight[i]) + "'\n")
f.write(" [../]\n")
for i in range(20):
f.write(" [./massfrac_" + var_name[i] + "_auxk]\n")
f.write(" type = ParsedAux\n")
f.write(" args = 'transported_mass transported_" + var_name[i] + "'\n")
f.write(" variable = massfrac_" + var_name[i] + "\n")
f.write(
" function = '"
+ str(mol_weight[i])
+ " * transported_"
+ var_name[i]
+ " / transported_mass'\n"
)
f.write(" [../]\n")
for mineral in all_minerals:
f.write(" [./dumped_" + mineral + "_auxk]\n")
f.write(" type = GeochemistryQuantityAux\n")
f.write(" variable = dumped_" + mineral + "\n")
f.write(" species = " + mineral + "\n")
f.write(" quantity = moles_dumped\n [../]\n")
f.write("[]\n")
f.write("\n")
f.write("[Postprocessors]\n")
for mineral in all_minerals:
f.write(" [./cumulative_moles_precipitated_" + mineral + "]\n")
f.write(" type = PointValue\n")
f.write(" variable = dumped_" + mineral + "\n [../]\n")
f.write(
" [./production_temperature]\n type = PointValue\n variable = production_temperature\n [../]\n [./mass_heated_this_timestep]\n type = PointValue\n variable = transported_mass\n [../]\n"
)
f.write("[]\n")
f.write("\n")
f.write("[Outputs]\n csv = true\n[]\n")
f.write("\n")
write_executioner(f)
f.write("\n")
f.write(
"[MultiApps]\n [./porous_flow_sim]\n type = TransientMultiApp\n input_files = "
+ porous_flow_filename
+ "\n execute_on = 'timestep_end'\n [../]\n[]\n"
)
f.write("\n")
f.write("[Transfers]\n")
f.write(
" [./injection_T]\n type = MultiAppNearestNodeTransfer\n direction = TO_MULTIAPP\n multi_app = porous_flow_sim\n fixed_meshes = true\n source_variable = 'solution_temperature'\n variable = 'injection_temperature'\n [../]\n"
)
for i in range(20):
f.write(" [./injection_" + var_name[i] + "]\n")
f.write(
" type = MultiAppNearestNodeTransfer\n direction = TO_MULTIAPP\n multi_app = porous_flow_sim\n fixed_meshes = true\n"
)
f.write(" source_variable = 'massfrac_" + var_name[i] + "'\n")
f.write(" variable = 'injection_rate_massfrac_" + var_name[i] + "'\n")
f.write(" [../]\n")
f.write("\n")
f.write(
" [./production_T]\n type = MultiAppPostprocessorInterpolationTransfer\n direction = FROM_MULTIAPP\n multi_app = porous_flow_sim\n postprocessor = production_temperature\n variable = production_temperature\n [../]\n"
)
for i in range(20):
f.write(" [./production_" + var_name[i] + "]\n")
f.write(
" type = MultiAppPostprocessorInterpolationTransfer\n direction = FROM_MULTIAPP\n multi_app = porous_flow_sim\n"
)
f.write(" postprocessor = mole_rate_" + var_name[i] + "_produced\n")
f.write(" variable = production_rate_" + var_name[i] + "\n")
f.write(" [../]\n")
f.write("[]\n")
f.close()
Results: precipitates in the heat exchanger
Precipitates form in the heat exchanger as the formation water is heated from the production temperature to 160C. Since the production temperature is more-or-less constant around 92C, because the simulation is not run long enough for significant thermal-breakthrough to occur, the precipitation rate is more-or-less constant. The results are shown in Figure 3. The results are very similar to the case in which a parcel of formation water is heated — see Figure 2 — with the greatest problem being Anhydrite. Note that this simulation heats the formation water, removing any precipitates formed and preventing any subsequent dissolution if any may occur, so it is a "worst-case" scenario.
Figure 3: Degree of scaling precipitate expected in the heat exchanger according to the full exchanger-porous_flow-aquifer_geochemistry model.
Results: mineralogy change around the injection well
Minerals precipitate and dissolve around the injection well as shown in Figure 4 and Figure 5. By volume, Illite and Kaolinite undergo most dissolution: indeed, 100% of these minerals are dissolved. In contrast, both K-feldspar and Quartz precipitate. Because the volume of dissolution exceeds the precipitated volume, the porosity increases marginally from its original value of 0.1.
Figure 4: Absolute change of mineral volume around the injection well.
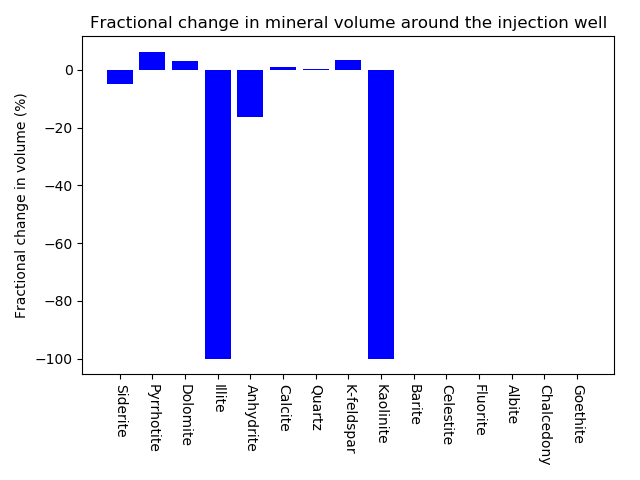
Figure 5: Relative change of mineral volume around the injection well.
Results: 3D contours
Figure 6 shows the results after 90 days of simulation. The input files producing these results employed a fine mesh and set remove_all_extrapolated_secondary_species = true in the GeochemicalModelDefinition to ensure convergence.
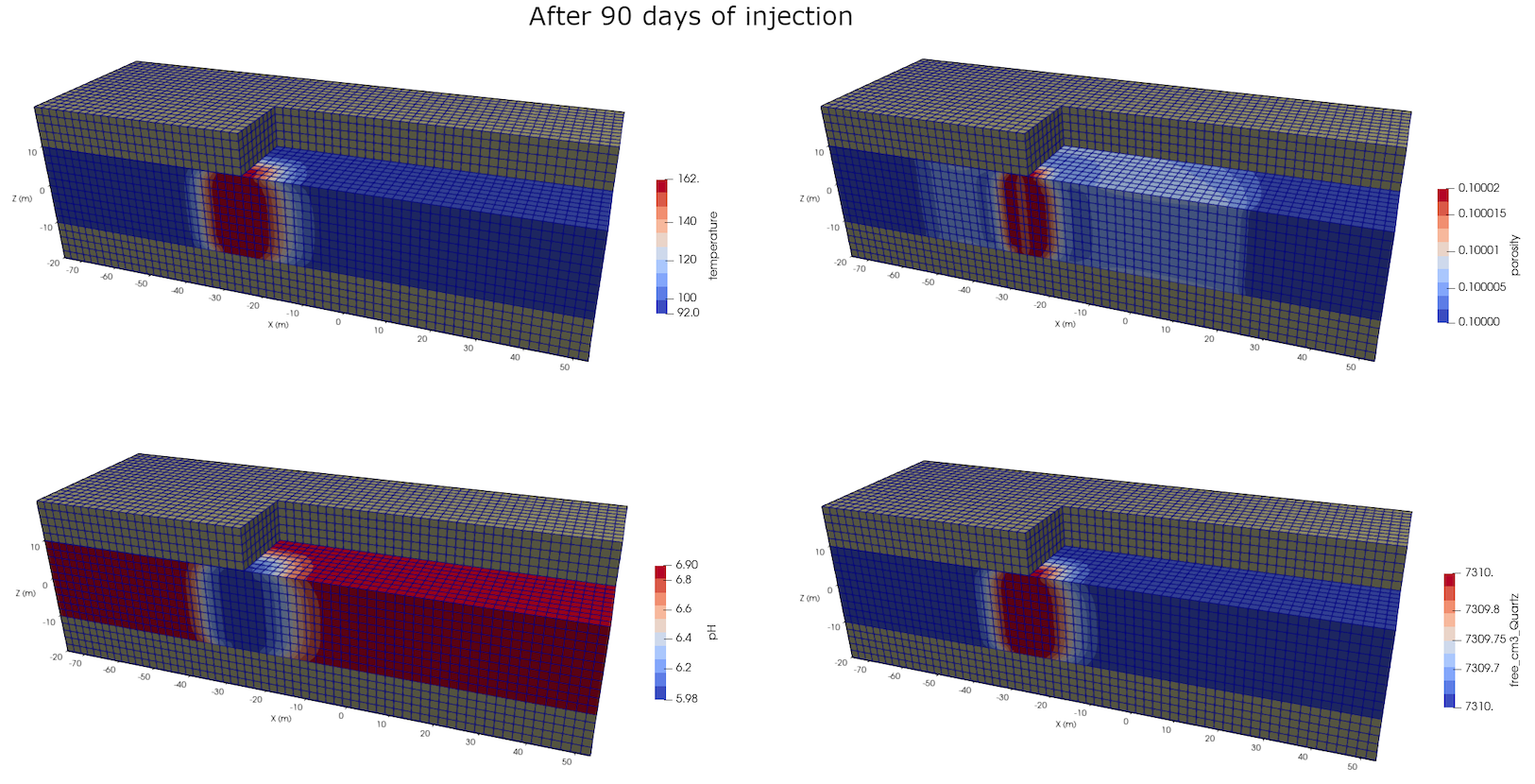
Figure 6: Temperature, porosity, pH and free volume of Quartz after 90 days of injection in the 3D coupled model.
Efficiencies
A related page explores the memory consumption, the impact of linear solver choices and the compute-time scaling with the number of processors of this problem.