Exploring the impact of pH on sorption
Section 14.3 of Bethke (2007) describes how to explore the impact of pH on sorption.
This page builds upon the surface complexation example in which sorption to the ferric hydroxide mineral Fe(OH) was studied. In that example, agreement between Bethke (2007), the Geochemists Workbench software, and the geochemistry module was demonstrated at pH 4 and 8. On this page, the pH is varied between 4 and 12, and amount of sorption of each species is plotted.
The surface complexation example included lead, mercury and iron complexes, but for simplicity this example only include iron complexes. The model definition therefore reads:
[UserObjects<<<{"href": "../../../syntax/UserObjects/index.html"}>>>]
[definition]
type = GeochemicalModelDefinition<<<{"description": "User object that parses a geochemical database file, and only retains information relevant to the current geochemical model", "href": "../../../source/userobjects/GeochemicalModelDefinition.html"}>>>
database_file<<<{"description": "The name of the geochemical database file"}>>> = "../../database/ferric_hydroxide_sorption.json"
basis_species<<<{"description": "A list of basis components relevant to the aqueous-equilibrium problem. H2O must appear first in this list. These components must be chosen from the 'basis species' in the database, the sorbing sites (if any) and the decoupled redox states that are in disequilibrium (if any)."}>>> = "H2O H+ Na+ Cl- Fe+++ >(s)FeOH >(w)FeOH"
equilibrium_minerals<<<{"description": "A list of minerals that are in equilibrium with the aqueous solution. All members of this list must be in the 'minerals' section of the database file"}>>> = "Fe(OH)3(ppd)"
piecewise_linear_interpolation<<<{"description": "If true then use a piecewise-linear interpolation of logK and Debye-Huckel parameters, regardless of the interpolation type specified in the database file. This can be useful for comparing with results using other geochemistry software"}>>> = true # for comparison with GWB
[]
[]The TimeDependentReactionSolver defines the initial composition as well as how to vary the pH with time via the controlled_activity inputs:
[TimeDependentReactionSolver<<<{"href": "../../../syntax/TimeDependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = "Fe+++"
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = "Fe(OH)3(ppd)"
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = "Cl-"
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = "H2O H+ Na+ Cl- Fe(OH)3(ppd) >(s)FeOH >(w)FeOH"
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = " 1.0 -4 0.1 0.1 9.3573E-3 4.6786E-5 1.87145E-3"
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = "kg_solvent_water log10activity bulk_composition bulk_composition free_mineral bulk_composition bulk_composition"
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = "kg dimensionless moles moles moles moles moles"
controlled_activity_name<<<{"description": "The names of the species that have their activity or fugacity constrained. There should be an equal number of these names as values given in controlled_activity_value. NOTE: if these species are not in the basis, or they do not have an activity (or fugacity) constraint then their activity cannot be controlled: in this case MOOSE will ignore the value you prescribe in controlled_activity_value."}>>> = "H+"
controlled_activity_value<<<{"description": "Values of the activity or fugacity of the species in controlled_activity_name list. These should always be positive"}>>> = set_aH
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 0 # not needed in this simple problem
stoichiometric_ionic_str_using_Cl_only<<<{"description": "If set to true, the stoichiometric ionic strength will be set equal to Cl- molality (or max_ionic_strength if the Cl- molality is too big). This flag overrides ionic_str_using_basis_molality_only"}>>> = true # for comparison with GWB
abs_tol<<<{"description": "If the residual of the algebraic system (measured in mol) is lower than this value, the Newton process (that finds the aqueous configuration) is deemed to have converged"}>>> = 1E-14
execute_console_output_on<<<{"description": "When to execute the geochemistry console output"}>>> = '' # only CSV output needed for this example
[]The set_aH value is defined via a FunctionAux in the following way:
[AuxVariables<<<{"href": "../../../syntax/AuxVariables/index.html"}>>>]
[set_aH]
[]
[]
[AuxKernels<<<{"href": "../../../syntax/AuxKernels/index.html"}>>>]
[set_aH]
type = FunctionAux<<<{"description": "Auxiliary Kernel that creates and updates a field variable by sampling a function through space and time.", "href": "../../../source/auxkernels/FunctionAux.html"}>>>
variable<<<{"description": "The name of the variable that this object applies to"}>>> = set_aH
function<<<{"description": "The function to use as the value"}>>> = '10^(-4-t)'
execute_on<<<{"description": "The list of flag(s) indicating when this object should be executed. For a description of each flag, see https://mooseframework.inl.gov/source/interfaces/SetupInterface.html."}>>> = timestep_begin # so the correct value is provided to the reactor
[]
[]and the timestepping is defined in the executioner:
[Executioner<<<{"href": "../../../syntax/Executioner/index.html"}>>>]
type = Transient
start_time = -0.25
dt = 0.25
end_time = 8
[]In this situation, the start_time is before when the system closes and the time-dependency begins, just so the results at can be simply recorded. Finally, the quantities of interest are recorded into Postprocessors using the AuxVariables that are automatically included in the simulation by the TimeDependentReactionSolver
[Postprocessors<<<{"href": "../../../syntax/Postprocessors/index.html"}>>>]
[pH]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'pH'
[]
[molal_>wFeOH2+]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(w)FeOH2+'
[]
[molal_>wFeOH]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(w)FeOH'
[]
[molal_>wFeO-]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(w)FeO-'
[]
[molal_>sFeOH2+]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(s)FeOH2+'
[]
[molal_>sFeOH]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(s)FeOH'
[]
[molal_>sFeO-]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_>(s)FeO-'
[]
[potential]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'surface_potential_Fe(OH)3(ppd)'
[]
[]Bethke (2007) presents results in Figures 14.8 and 14.9 (bold line only, since the fine line shows a different type of aqueous solution). The results are faithfully reproduced by the geochemistry module as shown in the figures below.
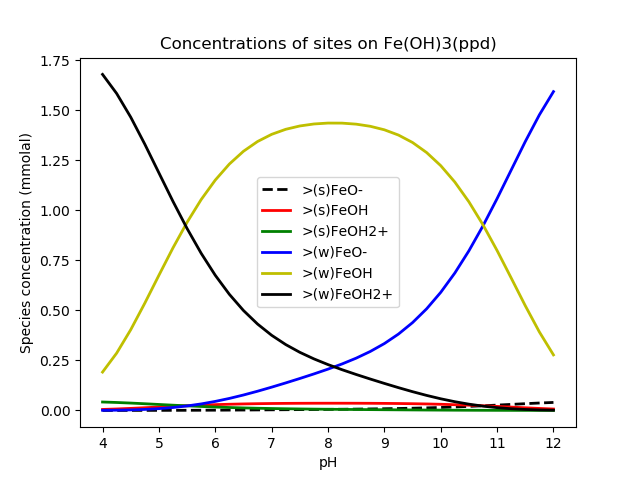
Figure 1: Concentrations of sites on a ferric oxide surface. Compare with Bethke's Figure 14.8
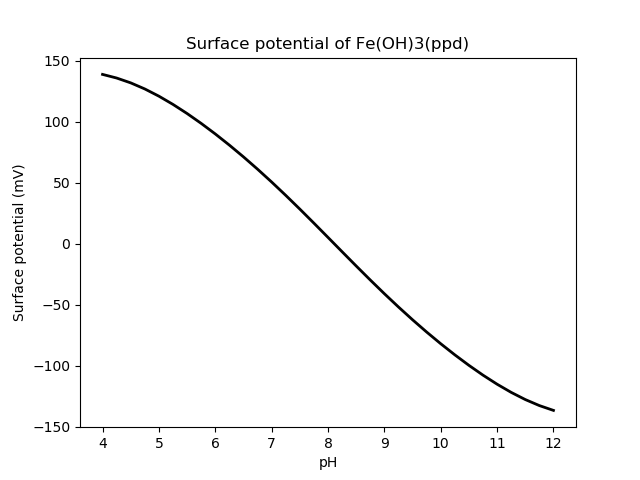
Figure 2: Surface potential of ferric oxide. Compare with Bethke's Figure 14.9
References
- Craig M. Bethke.
Geochemical and Biogeochemical Reaction Modeling.
Cambridge University Press, 2 edition, 2007.
doi:10.1017/CBO9780511619670.[Export]