Dumping minerals and then adding chemicals
Chapter 13.3 of Bethke (2007) describes a "dump" process, whereby a mineral is added to the initial solution, equilibrium sought, then any undissolved mineral is removed. In the code, this is achieved by:
subtracting from ;
then setting ;
setting the mole numbers of any surface components to zero, , as well as the molalities of unoccupied surface sites, and adsorbed species, ;
finally, swapping an appropriate aqueous species into the basis in place of .
After this process, other chemicals can be added to this system, and/or the temperature varied, and/or the pH varied, etc.
Section 15.2 of Bethke (2007) provides an example of this "dump" process. Assume:
the mineral calcite is used in the basis instead of HCO, and that the free mole number of the calcite is 0.2708 (corresponding to a free volume of calcite is cm);
the pH is initially 8;
the concentration of Na is 100mmolal;
the concentration of Ca is 10mmolal;
charge balance is enforced on Cl.
MOOSE input file
The GeochemicalModelDefinition defines the basis species, the equilibrium mineral and gas of interest in this problem, as well as using the piecewise_linear_interpolation flag for accurate comparisons with the Geochemists Workbench software:
[UserObjects<<<{"href": "../../../syntax/UserObjects/index.html"}>>>]
[definition]
type = GeochemicalModelDefinition<<<{"description": "User object that parses a geochemical database file, and only retains information relevant to the current geochemical model", "href": "../../../source/userobjects/GeochemicalModelDefinition.html"}>>>
database_file<<<{"description": "The name of the geochemical database file"}>>> = "../../../database/moose_geochemdb.json"
basis_species<<<{"description": "A list of basis components relevant to the aqueous-equilibrium problem. H2O must appear first in this list. These components must be chosen from the 'basis species' in the database, the sorbing sites (if any) and the decoupled redox states that are in disequilibrium (if any)."}>>> = "H2O H+ Na+ Cl- Ca++ HCO3-"
equilibrium_minerals<<<{"description": "A list of minerals that are in equilibrium with the aqueous solution. All members of this list must be in the 'minerals' section of the database file"}>>> = "Calcite"
equilibrium_gases<<<{"description": "A list of gases that are in equilibrium with the aqueous solution and can have their fugacities fixed, at least for some time and spatial location. All members of this list must be in the 'gas' section of the database file"}>>> = "CO2(g)"
piecewise_linear_interpolation<<<{"description": "If true then use a piecewise-linear interpolation of logK and Debye-Huckel parameters, regardless of the interpolation type specified in the database file. This can be useful for comparing with results using other geochemistry software"}>>> = true # for comparison with GWB
[]
[]The TimeDependentReactionSolver defines:
the swaps;
the initial bulk mole number of the aqueous species in the current basis, the free mole number of Calcite and the pH (via the H
activity);that the system is closed at (the default), so that after this time no more Calcite can be added to the system to maintain its free mole number;
that at the activity constraint is removed on H so that the pH can vary;
that HCl is added at a rate of 0.001mol/s;
using
mode = 1, that Calcite is dumped from the system after each time-step (calcite never precipitates in this example, so the dumping actually only happens at the first time-step).
[TimeDependentReactionSolver<<<{"href": "../../../syntax/TimeDependentReactionSolver/index.html"}>>>]
model_definition<<<{"description": "The name of the GeochemicalModelDefinition user object (you must create this UserObject yourself)"}>>> = definition
geochemistry_reactor_name<<<{"description": "The name that will be given to the GeochemistryReactor UserObject built by this action"}>>> = reactor
swap_out_of_basis<<<{"description": "Species that should be removed from the model_definition's basis and be replaced with the swap_into_basis species"}>>> = "HCO3-"
swap_into_basis<<<{"description": "Species that should be removed from the model_definition's equilibrium species list and added to the basis. There must be the same number of species in swap_out_of_basis and swap_into_basis. These swaps are performed before any other computations during the initial problem setup. If this list contains more than one species, the swapping is performed one-by-one, starting with the first pair (swap_out_of_basis[0] and swap_into_basis[0]), then the next pair, etc"}>>> = "Calcite"
charge_balance_species<<<{"description": "Charge balance will be enforced on this basis species. This means that its bulk mole number may be changed from the initial value you provide in order to ensure charge neutrality. After the initial swaps have been performed, this must be in the basis, and it must be provided with a bulk_composition constraint_meaning."}>>> = "Cl-"
constraint_species<<<{"description": "Names of the species that have their values fixed to constraint_value with meaning constraint_meaning. All basis species (after swap_into_basis and swap_out_of_basis) must be provided with exactly one constraint. These constraints are used to compute the configuration during the initial problem setup, and in time-dependent simulations they may be modified as time progresses."}>>> = "H2O Calcite Ca++ Na+ Cl- H+"
constraint_value<<<{"description": "Numerical value of the containts on constraint_species"}>>> = " 1.0 10 0.01 0.1 0.11 -8"
constraint_meaning<<<{"description": "Meanings of the numerical values given in constraint_value. kg_solvent_water: can only be applied to H2O and units must be kg. bulk_composition: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species but not in kinetic species, and units must be moles or mass (kg, g, etc). bulk_composition_with_kinetic: can be applied to all non-gas species, and represents the total amount of the basis species contained as free species as well as the amount found in secondary species and in kinetic species, and units must be moles or mass (kg, g, etc). free_concentration: can be applied to all basis species that are not gas and not H2O and not mineral, and represents the total amount of the basis species existing freely (not as secondary species) within the solution, and units must be molal or mass_per_kg_solvent. free_mineral: can be applied to all mineral basis species, and represents the total amount of the mineral existing freely (precipitated) within the solution, and units must be moles, mass or cm3. activity and log10activity: can be applied to basis species that are not gas and not mineral and not sorbing sites, and represents the activity of the basis species (recall pH = -log10activity), and units must be dimensionless. fugacity and log10fugacity: can be applied to gases, and units must be dimensionless"}>>> = "kg_solvent_water free_mineral bulk_composition bulk_composition bulk_composition log10activity"
constraint_unit<<<{"description": "Units of the numerical values given in constraint_value. Dimensionless: should only be used for activity or fugacity constraints. Moles: mole number. Molal: moles per kg solvent water. kg: kilograms. g: grams. mg: milligrams. ug: micrograms. kg_per_kg_solvent: kilograms per kg solvent water. g_per_kg_solvent: grams per kg solvent water. mg_per_kg_solvent: milligrams per kg solvent water. ug_per_kg_solvent: micrograms per kg solvent water. cm3: cubic centimeters"}>>> = " kg cm3 moles moles moles dimensionless"
ramp_max_ionic_strength_initial<<<{"description": "The number of iterations over which to progressively increase the maximum ionic strength (from zero to max_ionic_strength) during the initial equilibration. Increasing this can help in convergence of the Newton process, at the cost of spending more time finding the aqueous configuration."}>>> = 10
remove_fixed_activity_name<<<{"description": "The name of the species that should have their activity or fugacity constraint removed at time given in remove_fixed_activity_time. There should be an equal number of these names as times given in remove_fixed_activity_time. Each of these must be in the basis and have an activity or fugacity constraint"}>>> = 'H+'
remove_fixed_activity_time<<<{"description": "The times at which the species in remove_fixed_activity_name should have their activity or fugacity constraint removed."}>>> = 0
source_species_names<<<{"description": "The name of the species that are added at rates given in source_species_rates. There must be an equal number of these as source_species_rates."}>>> = 'HCl'
source_species_rates<<<{"description": "Rates, in mols/time_unit, of addition of the species with names given in source_species_names. A negative value corresponds to removing a species: be careful that you don't cause negative mass problems!"}>>> = 1E-3
mode<<<{"description": "This may vary temporally. If mode=1 then 'dump' mode is used, which means all non-kinetic mineral masses are removed from the system before the equilibrium solution is sought (ie, removal occurs at the beginning of the time step). If mode=2 then 'flow-through' mode is used, which means all mineral masses are removed from the system after it the equilbrium solution has been found (ie, at the end of a time step). If mode=3 then 'flush' mode is used, then before the equilibrium solution is sought (ie, at the start of a time step) water+species is removed from the system at the same rate as pure water + non-mineral solutes are entering the system (specified in source_species_rates). If mode=4 then 'heat-exchanger' mode is used, which means the entire current aqueous solution is removed, then the source_species are added, then the temperature is set to 'cold_temperature', the system is solved and any precipitated minerals are removed, then the temperature is set to 'temperature', the system re-solved and any precipitated minerals are removed. If mode is any other number, no special mode is active (the system simply responds to the source_species_rates, controlled_activity_value, etc)."}>>> = 1 # in this case, Calcite never re-precipitates, so never need to turn the dump option off
stoichiometric_ionic_str_using_Cl_only<<<{"description": "If set to true, the stoichiometric ionic strength will be set equal to Cl- molality (or max_ionic_strength if the Cl- molality is too big). This flag overrides ionic_str_using_basis_molality_only"}>>> = true # for comparison with GWB
execute_console_output_on<<<{"description": "When to execute the geochemistry console output"}>>> = '' # only CSV output for this test
[]The Executioner provides meaning to time, in particular that 100s is used, so a total of 0.1mol of HCl is added:
[Executioner<<<{"href": "../../../syntax/Executioner/index.html"}>>>]
type = Transient
dt = 10
end_time = 100
[]The results presented below were run with a time-step of 1 to improve the appearance of the graphs.
A set of Postprocessors record the desired information using AuxVariables automatically added by the TimeDependentReactionSolver:
[Postprocessors<<<{"href": "../../../syntax/Postprocessors/index.html"}>>>]
[cm3_Calcite]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'free_cm3_Calcite'
[]
[pH]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'pH'
[]
[molal_CO2aq]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_CO2(aq)'
[]
[molal_CaCl+]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_CaCl+'
[]
[molal_HCO3-]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_HCO3-'
[]
[molal_Ca++]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'molal_Ca++'
[]
[fugacity_CO2]
type = PointValue<<<{"description": "Compute the value of a variable at a specified location", "href": "../../../source/postprocessors/PointValue.html"}>>>
point<<<{"description": "The physical point where the solution will be evaluated."}>>> = '0 0 0'
variable<<<{"description": "The name of the variable that this postprocessor operates on."}>>> = 'activity_CO2(g)'
[]
[]GWB input file
The equivalent Geochemists Workbench file is
# React script that is equivalent to calcite_dumping.i
data = thermo.tdat verify
conductivity = conductivity-USGS.dat
temperature = 25 C
H2O = 1 free kg
H+ = 8 pH
Na+ = 0.1 mol
Ca++ = 0.01024844 mol
Cl- = 0.12 mol
balance on Cl-
swap Calcite for HCO3-
Calcite = 0.2708 free mol
suppress all
unsuppress Calcite
printout species = long
epsilon = 1e-13
dump
react 0.1 mol of HCl
go
Note the slightly increased Ca bulk composition: this is explained here.
Results
Two cases are run:
The system is brought to equilibrium and then the calcite is "dumped" using
mode = 1in the input file. After this, 100mmol of HCl is added to the system.The system is brought to equilibrium and but the calcite is not "dumped" using
mode = 0in the input file. After this, 100mmol of HCl is added to the system.
Bethke (2007) presents the results in Figures 15.6 and 15.7. Both GWB and the geochemistry module produce the same results, as shown below.
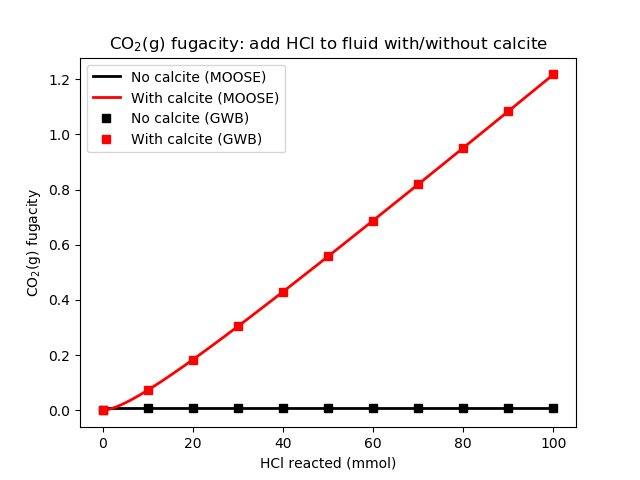
Figure 1: CO2(g) fugacity as HCl is added to the fluid. Compare with Bethke's Figure 15.6
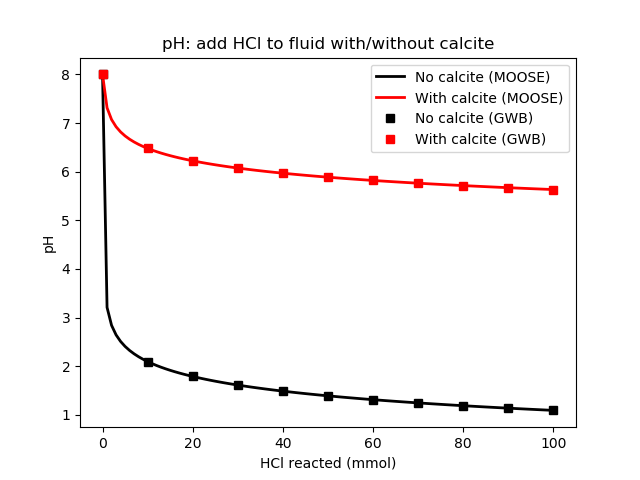
Figure 2: pH as HCl is added to the fluid. Compare with Bethke's Figure 15.6
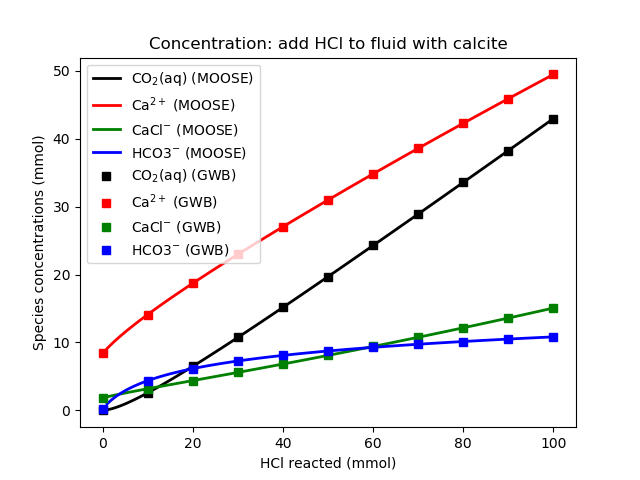
Figure 3: Species concentrations as HCl is added to the fluid. Compare with Bethke's Figure 15.7
References
- Craig M. Bethke.
Geochemical and Biogeochemical Reaction Modeling.
Cambridge University Press, 2 edition, 2007.
doi:10.1017/CBO9780511619670.[Export]